
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Almost half of new molecular entities approved in 2011 considered significant therapeutic advances
New molecular entities (NMEs) for calendar year 2011 number 30, considerably above the average of 23 during the past 10 years. Notably among this list are 8 drugs for cancer; 10 targeting orphan diseases; 2 each for hepatitis C and chronic obstructive pulmonary disease (COPD); and 3 anticoagulants.



Key Points
Abstract
Of the 30 new molecular entities (NMEs) approved last year by FDA, almost half of the drugs were considered to be significant therapeutic advances over existing therapies for hepatitis C, stroke, and kidney transplant. The majority of the approved drugs received "priority review," giving FDA a 6-month goal to complete its evaluation for safety and efficacy. About two-thirds of the new approvals did not require giving additional information to FDA, so they were completed in a single review cycle. Approximately 70% of the new drugs were approved in the United States before any other country, including the European Union. (Formulary. 2012; 47:26-38.)
New molecular entities (NMEs) for calendar year 2011 number 30, considerably above the average of 23 during the past 10 years. Notably among this list are 8 drugs for cancer; 10 targeting orphan diseases; 2 each for hepatitis C and chronic obstructive pulmonary disease (COPD); and 3 anticoagulants. Four of these NMEs also fall under biologics license application (BLA) approvals, 2 of which are for cancer, 1 for systemic lupus erythematosus, and 1 for kidney transplant rejection.
"Some new drugs represent major breakthroughs in therapy while some turn out to be "me-too" drugs, offering no or only minor benefits compared to drugs that may already be available," says William Simonson, independent consultant pharmacist and senior research professor of pharmacy practice at Oregon State University.
NMEs are drugs that use an active ingredient that has never before been marketed in the United States in any form. They include drugs approved by FDA's Center for Drug Evaluation and Research and FDA's Center for Biologics Evaluation and Research:
Realizing a gap in treatment for a variety of conditions, FDA quickly approved almost every drug on or before the review time targets agreed to with industry under the 1992 Prescription Drug User Fee Act (PDUFA). PDUFA allows FDA to charge drug manufacturers a substantial application fee for funding drug approvals, while in turn FDA must meet certain performance benchmarks primarily associated with the speed of review.
"What makes 2011 stand out is the number of NME drugs approved in the first half of the year [18]-greater than during the previous year," says Brian Kolling, PharmD, senior director, pipeline and trend forecasting, Part D for OptumRX, a pharmacy benefit management company headquartered in Cypress, Calif. "Although some approvals depend on when the drug is filed, if 2011 had continued at the same pace as it started out, this would have been one of the most productive years in drug development."
"This year represents the beginning of a very innovative trend in the marketplace," says Randy Vogenberg, PharmD, principal, Institute for Integrated Healthcare, Sharon, Mass.
"There is an improved understanding of treatment strategies for better managing disease through a patient's lifetime. Combination therapies for HIV and cancer are indications of how well we have learned to look for multiple pathways and markers when a patient is in trouble with chronic conditions," he says.
Vogenberg also points out that some orphan drugs have entered the marketplace this year that have been progressing in the pipeline for the past 8 to 10 years. "This is just the tip of the iceberg," he says. On the other hand, he acknowledges that orphan drugs will never become blockbusters because they target such a small population.
The following list of NMEs/BLAs for calendar year 2011 is outlined by indication. Accompanying Tables 1 and 2 highlight the drugs' generic and brand names, indication, manufacturer, and date of approval. Much of the information was gleaned from these FDA website locations: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm254242.htm, http://www.accessdata.fda.gov/scripts/cder/drugsatfda/, and http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/BiologicalApprovalsbyYear/ucm242993.htm,
CANCER

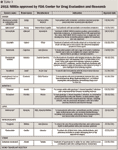
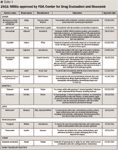
A new drug to treat metastatic, castration-resistant prostate cancer in patients who have received prior docetaxel therapy, abiraterone acetate (Zytiga) is a pill that targets the protein referred to as cytochrome P450 17A1 (CYP17A1), which plays an important role in the production of testosterone. The drug decreases the production of this hormone that stimulates cancer cell growth. Approved ahead of its regulatory goal date, it is administered in combination with prednisone.




Ipilimumab is a monoclonal antibody that blocks a molecule that may play a part in slowing down or turning off the body's immune system, limiting its ability to fight off cancerous cells. Delivered intravenously, ipilimumab is expected to recognize, target, and attack melanoma tumors. The American Cancer Society has estimated that 68,130 new cases of melanoma were diagnosed in 2010 in the United States.
A single international study of 676 patients with metastatic or inoperable melanoma who were no longer responding to FDA-approved treatments for the disease weighed ipilimumab plus an experimental tumor vaccine (gp100), ipilimumab alone, or the vaccine alone. Survival rates using ipilimumab and the vaccine or ipilimumab alone outperformed the last alternative-10 months compared to 6.5 months. The major drawback of the drug is a fatal autoimmune reaction seen in 12.9% of patients. The drug is being investigated to treat other cancers.
FDA approved the personalized drug therapies vemurafenib (Zelboraf) and crizotinib (Xalkori) in 2011. These therapies target patients with specific genetic mutations who have been previously tested for the abnormalities through a companion genetic test.
Vemurafenib treats patients with late-stage metastatic or inoperable melanoma, the most dangerous form of skin cancer; crizotinib addresses late-stage, non-small-cell lung cancer (NSCLC). Both have been designated as orphan drugs. Vemurafenib has been FDA approved with a first-of-a-kind genetic test called cobas 4800 BRAF V600 mutation test to detect which patients have the genetic mutation BRAF V600E, which vemurafenib addresses by blocking its function.
The median survival (length of time a patient lives after treatment) of patients receiving vemurafenib has not been reached (77% still living), whereas the median survival for those who received dacarbazine-another anticancer therapy tested in a trial against vemurafenib-was 8 months (64% still living). Vemurafenib is being approved with a medication guide to inform healthcare professionals and patients of vemurafenib's potential risks. A single international trial of 675 subjects tested patients who had not received prior treatment for melanoma.
Vemurafenib is the second new cancer drug (after ipilimumab) ever approved by FDA that has demonstrated an improvement in overall survival. Vemurafenib is being tested in combination with ipilimumab and as a treatment for other cancers. Projected sales are expected to be $569 million by 2016.
Crizotinib treats certain patients with NSCLC that express the abnormal anaplastic lymphoma kinase (ALK) gene, which causes cancer development and growth. About 1% to 7% of those with NSCLC have the gene. Crizotinib has been approved with a companion diagnostic test to determine if a patient has an abnormal ALK gene. Called the Vysis ALK Break-Apart FISH Probe Kit, it is the first-of-a-kind genetic test.
Crizotinib's side effects include vision disorders, nausea, vomiting, and swelling, as well as a more severe condition, pneumonitis. The drug took only 5 years from first-in-human phase 1 studies to FDA approval. It is taken orally twice daily.
Sherry Andes, senior drug information specialist, SCX Health Solutions based in Lisle, Ill., suggests that there may be some potential barriers to adopting the companion diagnostic for crizotinib, including concerns about FISH-based assays, the amount of tissue required for the assay, and the need to re-biopsy a tumor.
The first drug to treat medullary thyroid cancer, which represents 3% to 5% of thyroid cancers, is vandetanib (Caprelsa). The cancer can occur spontaneously and is an orphan disease, as well as one of the rarest forms of thyroid cancer.
A single, randomized international trial of 331 patients showed that patients who received vandetanib had a median progression-free survival of at least 22.6 months compared to 16.4 months for the placebo. The drug was approved with a Risk Evaluation and Mitigation Strategy (REMS) to notify healthcare professionals of the risk of heart-related problems. The drug is delivered orally on a daily basis.
"Health professionals and patients have to weigh the advantages that new drugs offer against the potential risk of side effects, drug interactions, or other complications," Simonson says.
Common side effects include diarrhea, rash, nausea, hypertension, headache, and fatigue.
Another new oncology drug, brentuximab vedotin (Adcetris), targets patients with Hodgkin lymphoma whose disease has worsened after autologous stem cell transplant or after failure of at least 2 chemotherapy treatments for which a transplant is not an option. It is the first new FDA-approved drug to treat Hodgkin lymphoma since 1977. The National Cancer Institute has estimated that 8830 new cases will be diagnosed in the United States in 2011, with 1300 dying from the disease.
Brentuximab vedotin, which also treats systemic anaplastic large cell lymphoma (ALCL), is an antibody conjugate enabling the antibody to hit a target on lymphoma cells named CD30. The drug also helps patients with ALCL, whose disease has progressed despite a prior chemotherapy treatment.
A single-arm trial of only 102 patients with Hodgkin lymphoma resulted in an objective response rate of 73% who experienced complete or partial cancer shrinkage or disappearance after treatment; on average they responded for 6.7 months. The single-arm, 58-person trial for ALCL showed an 86% partial or complete response for an average of 12.6 months. Adverse effects include neutropenia, nerve damage, low blood platelet counts (thrombocytopenia), and nausea.
Ruxolitinib phosphate (Jakafi) is the first drug approved to specifically treat patients with the rare bone marrow disease myelofibrosis, in which the bone marrow is replaced by scar tissue causing organs such as the liver and spleen to produce blood cells. The disease is characterized by an enlarged spleen, anemia, decreased white blood cells and platelets, abdominal discomfort, fatigue, and muscle and bone pain. The most serious side effects include fatigue, dizziness, headache, and nausea.
"Prior to the approval of ruxolitinib, patients with myelofibrosis who are not candidates for an allergenic bone marrow transplant were treated with medications, such as hydroxyurea, chemotherapy drugs, or glucocorticoids. Ruxolitinib is the first in a new class of drugs known as Janus associated kinase (JAK)," explains Aimee Tharaldson, senior clinical consultant, Emerging Therapeutics at Express Scripts in Bloomington, Minn.
The twice-a-day tablet prevents enzymes that help regulate blood and immunologic functioning. It has been designated as an orphan drug; myelofibrosis affects approximately 18,000 people in the United States.
In 2 randomized phase 3 studies, patients were selected to receive treatment with either ruxolitinib, placebo, or the best available therapy (hydroxyurea, a chemotherapy agent, or glucocorticoids). A greater percentage of patients receiving ruxolitinib experienced more than a 35% reduction in spleen volume when compared to patients receiving placebo or best available therapy. Similarly, a greater proportion of patients receiving ruxolitinib saw more than a 50% reduction in their myelofibrosis-related symptoms than was the case in patients receiving placebo.
Kolling is optimistic that ruxolitinib with its efficacy will fulfill an unmet need for myelofibrosis, even though it will only treat a small number of individuals.
AsparaginaseErwinia chrysanthemi (Erwinaze) is the first and only approved drug to treat patients with acute lymphoblastic leukemia (ALL), who have developed an allergy to Escherichia coli-derived asparaginase and pegapargase chemotherapy drugs used for the condition. ALL is a type of cancer that causes bone marrow to produce too many lymphocytes, a type of white blood cell. Asparaginase Erwinia chrysanthemi received an orphan drug designation.
Two clinical trials indicated that patients needed to discontinue therapy due to allergic reactions; however, the trial that supports efficacy showed that all evaluable patients maintained the prespecified threshold for asparaginase activity-levels that correspond to better leukemia control and survival-at 48 and 72 hours after dosing.
HEPATITIS C
Both telaprevir (Incivek) and boceprevir (Victrelis) are new protease inhibitors to treat chronic hepatitis C virus (HCV) that causes inflammation of the liver. As many as 3 million people in the United States have hepatitis C. These drugs represent a new therapy for patients with the condition; they target chronic HCV genotype 1 infection.
Andes notes that telaprevir is generally regarded as the more potent of the drugs, which have not yet been tested head to head.
Boceprevir, approved 10 days before telaprevir, targets patients who have some liver function and have not been previously treated with a drug therapy for hepatitis or who have failed the treatment.
In a pair of phase 3 clinical trials with 1500 adult patients, two-thirds of those receiving boceprevir in combination with pegylated interferon and ribavirin-the latter being the standard of care-experienced significant increases in sustained virologic response (SVR), better than with each of the other drugs alone. Recommended dosage is 3 times daily with food and does not require dosage adjustment in patients with renal or hepatic impairment.
Standard treatment is 48 weeks of pegylated interferon and ribavirin; however, studies have shown this therapy to only achieve SVR in 44% of patients.
Telaprevir combined with peginterferon alfa-2a and ribavirin for the first 12 weeks of treatment has shown improved response in about 75% of patients. The combination also has shortened the treatment duration by half for those who have indicated improvement after the 12-week combination drug therapy.
Kolling points out that peginterferon was approved in 2001 and ribavirin in 1998 as the only treatment options; however, they may not cure hepatitis C. "Without a liver transplant, many patients had no other alternatives. With these 2 new drugs, in combination with interferon and ribavirin, these patients can be treated again," he says.
LUPUS
Belimumab (Benlysta) is the first new treatment for systemic lupus erythematosus, an autoimmune disorder characterized by exacerbations and remissions, in more than 50 years, and the first inhibitor designed to target B-lymphocyte stimulator (BLyS) protein. It received an orphan drug designation. The disease primarily affects women between the ages of 15 and 44 years and is often treated by nonsteroidal anti-inflammatory drugs and corticosteroids.
Belimumab modifies the proliferation of BLyS protein, which when elevated, can cause the production of cells that attack patients' blood vessels and organ systems, according to Tharaldson.
FDA put belimumab on a fast track priority review. Although its blockbuster potential seems high, Kolling is skeptical.
ANTICOAGULANTS
Two new anticoagulants were approved in July 2011-ticagrelor (Brilinta) and rivaroxaban (Xarelto), the latter of which follows in the wake of dabigatran (Pradaxa), the first oral anti-clotting drug used to prevent stroke in patients with atrial fibrillation. Approved in October 2010 dabigatran was the first oral anticoagulant to challenge warfarin that was developed in the 1950s. Warfarin has been the standard recommended drug for atrial fibrillation; however, it requires frequent monitoring, which is not necessary with rivaroxaban.
Facing off with clopidogrel (Plavix), the oral antiplatelet ticagrelor was approved to reduce cardiovascular death for patients with acute coronary syndrome that can reduce blood flow to the heart. It prevents the formation of new blood clots allowing blood to flow in the body and thus reducing the risk of another cardiovascular event. A boxed warning alerts patients that daily aspirin doses above 100 mg decrease effectiveness and that the drug increases the rate of bleeding.
Kolling says that although ticagrelor will fill a niche, it will be more expensive than clopidogrel, which goes off patent this year. At the same time, he believes that if patients are stable on clopidogrel, they will continue to take the medication. He also points out that ticagrelor is geared more toward patients with acute coronary syndrome; however, this may change as the drug is studied in additional settings.
Clinical trials show that ticagrelor is more effective than clopidogrel. It was approved with a REMS to help ensure that the drug's benefits outweigh its risks.
Andes adds that ticagrelor's primary disadvantages may be its twice-a-day dosing compared to once-daily doses of clopidogrel and prasugrel (Effient), an oral antiplatelet medication for patients who have undergone angioplasty, and its aspirin limitations.
Rivaroxaban is a novel oral factor Xa inhibitor taken once daily that prevents stroke in nonvalvular atrial fibrillation and reduces the risk of blood clots, deep vein thrombosis, and pulmonary embolism following hip or knee replacement surgery. Recommended dosing is 10 mg taken orally once a day for 12 days after knee replacement surgery and 35 days for hip replacements.
Clinical studies targeted the occurrence of venous thromboembolic events (VTE) after both kinds of surgeries. When compared to enoxaparin, about half as many knee replacement patients taking rivaroxaban experienced VTE, and for hip replacements, only 1.1% of those taking rivaroxaban did compared to 3.9% on enoxaparin. Bleeding is the most significant adverse effect.
According to a recent study published in the New England Journal of Medicine (2011;365:2002-2012), one-third of emergency room cases involved warfarin, which, when not monitored closely, can cause hemorrhaging.
Apixaban (Eliquis), another blood thinner used to prevent strokes, as well as systemic embolism, in those with atrial fibrillation, is expected to be approved in a priority review by FDA by March 28, 2012.
STROKE
Another drug that addresses stroke, azilsartan medoxomil (Edarbi), is not an anticoagulant but rather treats hypertension and prevents the action of the vasopressor hormone. Azilsartan medoxomil joins 7 other angiotensin II receptor blockers in the marketplace. Clinical studies indicate its performance exceeded valsartan and olmesartan, other FDA previously approved hypertension drugs.
The suggested dose for azilsartan medoxomil is 80 mg once daily and 40 mg for patients using high-dose diuretics to reduce salt content. Its side effects are similar to those from a placebo. The drug is not recommended for women who are pregnant.
KIDNEY TRANSPLANT REJECTION
Belatacept (Nulojix) is the first drug in a new class of primary immunosuppressants preventing acute organ rejection of transplanted kidneys in adults. Belatacept, delivered through a 30-minute intravenous infusion, works in tandem with other immunosuppressants, such as corticosteroids, to manage the immune system and ensure the new kidney is working.
More than 89,000 patients are on the waiting list for a kidney transplant in the United States, according to the US Department of Health and Human Services. Designated as a fast track drug, it carries a boxed warning because the drug increases the risk of a post-transplant lymphoproliferative disorder, a type of cancer that is more prevalent for transplant patients who have never been exposed to Epstein-Barr virus. Patients should only use the new drug if they have already been exposed.
Two open-label, randomized, multicenter, controlled phase 3 studies with 1200 patients compared belatacept with cyclosporine and showed that the former is safe and effective for organ rejection.
Bristol-Myers Squibb, the manufacturer, has estimated that 80% of the 17,000 patients receiving a kidney transplant will be candidates for the drug.
ANTISEIZURE MEDICATIONS
In 2011 the new antiseizure drugs, clobazam (Onfi) and ezogabine (Potiga) were approved. Clobazam is an oral benzodiazepine for adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in adults and children aged 2 years and older. Lennox-Gastaut is a severe form of epilepsy that causes debilitating seizures, including brief loss of muscle tone and consciousness, staring spells, pupil dilation, and stiffening of the body.
FDA granted an orphan drug designation to clobazam, which treats a disease suffered by fewer than 200,000 in the United States. It usually starts before age 4 and most children with the syndrome experience some degree of impaired intellectual functioning or information processing.
Two multicenter controlled studies of patients aged 2 years and older tested the drug for the amount of reduction in the weekly frequency of drop seizures resulting in a fall from the 4-week baseline period to the maintenance period. When added to ongoing seizure medication, clobazam indicated improved seizure control compared to a placebo and low-dose clobazam. Side effects include fever, constipation, cough, insomnia, irritability, and respiratory tract infection.
As an add-on drug, ezogabine treats seizures associated with epilepsy in adults, more specifically partial-onset seizures that are the most common type for epilepsy patients. Ezogabine is the first neuronal potassium channel opener for treating epilepsy, and although its mechanism of action is not fully understood, the drug may act as an anticonvulsant. Its use may cause urinary retention (which should be monitored), confusion, and occasional suicidal thoughts. A medication guide accompanies the prescription.
Andes points out that the drug ezogabine enters the marketplace against multiple generics and established branded products.
IRON OVERLOAD
Deferiprone (Ferriprox) addresses patients with iron overload due to blood transfusions in patients with thalassemia, a genetic blood disorder that causes anemia. Candidates for the drug also had an inadequate response to prior chelation therapy-standard therapy to treat iron overload that uses chemical agents to remove heavy metals from the body. Deferiprone is the first new FDA-approved treatment for this disorder since 2005.
The drug's success is based on a 20% reduction in serum ferritin, which was realized by 50% of patients in data from 12 clinical studies with 236 patients.
Although patients who took deferiprone may have experienced nausea, vomiting, and neutropenia, 2% developed a life-threatening decrease in white blood cells that fight infection.
COPD
Two new drugs address COPD-indacaterol maleate (Arcapta Neohaler) and roflumilast (Daliresp).
The inhalation powder indacaterol maleate is a long-term, once-daily maintenance bronchodilator that treats airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. It is a NME in the beta2-adrenergic agonist class of drugs that relaxes muscles around the lungs to prevent COPD symptoms. It is not prescribed to treat asthma or severe symptoms of COPD.
The drug's safety and efficacy were qualified in 6 clinical trials with 5474 patients aged 40 and older who had smoked at least a pack a day for 10 years and showed moderate-to-severe decreases in lung function. The drug is accompanied by a boxed warning that long-acting beta2-adrenergic agonists can increase the risk of asthma-related deaths.
A new class of drug for COPD, roflumilast is orally administered in a daily tablet that decreases the frequency of flare-ups and helps prevent worsening COPD symptoms, which include breathlessness, chronic cough, and excessive phlegm. Roflumilast oral treatment, a phosphodiesterase type 4 (PDE-4) inhibitor, inhibits an enzyme called PDE-4 and targets those with severe COPD, not patients with COPD that involves primary emphysema, sudden breathing problems, or who are younger than 18 years of age.
The drug's efficacy was demonstrated through a phase 3 clinical trial of more than 1500 patients aged 40 or older who had a history of COPD with chronic bronchitis and had suffered from symptoms during the 12 months prior to treatment.
EYES
Aflibercept (Eylea) has been approved to treat patients with wet (neovascular), age-related macular degeneration (AMD), a leading cause of vision loss and blindness in US citizens aged 60 years and older. The recommended dose for the eye injection is 2 mg monthly for the first 12 weeks, followed by 2 mg bimonthly. AMD gradually destroys a person's sharp, central vision.
This approval was based on the results of 2 phase 3 clinical studies. In these studies, which included a total of 2412 patients, aflibercept dosed every 8 weeks, following 3 initial monthly injections, was clinically equivalent to the standard of care, ranibizumab (Lucentis), dosed every 4 weeks. Measurement was measured by the primary end point of maintenance of visual acuity (<15 letters of vision loss on an eye chart) over 52 weeks. The most common adverse reactions reported in patients receiving aflibercept were conjunctival hemorrhage, eye pain, cataract, vitreous detachment, vitreous floaters, and increased intraocular pressure.
DIARRHEA
Fidaxomicin (Dificid) treats Clostridium difficile-associated diarrhea (CDAD), which can result in colitis and other serious intestinal conditions. C. difficile is a spore-forming, gram-positive anaerobic bacillus transmitted to humans via the fecal-oral route. The spores remain inactive until an event that eliminates or disrupts the gut flora, triggering CDAD.
Two antibiotics, vancomycin and metronidazole, have served as the most effective treatments, but an increased number of reports have shown that these drugs have failed or resulted in recurrences. Fidaxomicin is the first anti-infective drug agent against C. difficile to be approved by FDA to treat CDAD.
The recommended dose for fidaxomicin, a macrolide antibacterial drug, is 200 mg 2 times a day for 10 days with or without food. It should only be used to treat infections that are proven or highly suspected of being the result of C. difficile. Clinical trials indicated that fidaxomicin is superior to vancomycin, a common antibiotic, in sustained clinical response time.
Although the cost of fidaxomicin for the 10-day course is expected to exceed $2500, data presented at the recent American Society of Health-System Pharmacists' 46th midyear clinical meeting (New Orleans, December 4-8, 2011) demonstrate that the economic value of the drug to the US healthcare system meets or exceeds the price per day to treat CDAD compared to vancomycin, providing justification for its high price.
Vogenberg sees this niche drug as important for treating CDAD, which is common in long-term care institutions where the condition can easily spread among patients.
HIV
Used in conjunction with other anti-retroviral drugs treating HIV-1 in adults who have never had HIV therapy, rilpivirine hydrochloride (Edurant) belongs to a class of HIV drugs called nonnucleoside reverse transcriptase inhibitors. Taken once a day, rilpivirine blocks HIV viral replication.
Phase 3 clinical trials tested rilpivirine against efavirenz, both in combination with other drugs, and found them equally effective in lowering viral load after 48 weeks of treatment. Patients with higher viral loads at the onset of therapy were less likely to respond to rilpivirine. Side effects include depression, insomnia, headache, and rash.
Rilpivirine is not a cure for HIV; instead, patients must continue on HIV therapy and try to prevent HIV-related illnesses.
DIABETES MELLITUS
Type 2 diabetes, affecting between 90% and 95% of the 24 million diabetics in the United States, gets a boost with the approval of linagliptin (Tradjenta) tablets. It represents the third dipeptidyl peptidase 4 inhibitor in the US marketplace. According to IMS Institute for Informatics in Norwalk, Conn., linagliptin sales are projected at $50 million in 2011, $100 million in 2012, and $400 million in 2015.
When used with proper exercise and diet, the drug improves blood glucose control in adults. In 8 double-blind, placebo-controlled clinical studies of 3800 patients, it proved to control blood glucose better than the placebo. Side effects are upper respiratory infection, sore throat, muscle pain, headache, and stuffy or runny nose.
RESTLESS LEGS SYNDROME
A new drug has been approved for a non-life-threatening but disruptive neurologic disorder-restless legs syndrome (RLS)-that affects up to 10% of the population, according to the RLS Foundation. Known as gabapentin enacarbil (Horizant), this extended-release tablet is given once a day for moderate-to-severe RLS.
Its effectiveness was researched through a couple of 12-week clinical trials in adults, in which it performed better than a placebo in reducing symptoms. It is distributed with a medication guide, highlighting gabapentin enacarbil's ability to cause drowsiness and dizziness.
"This modified gabapentin prodrug is useful for RLS; however, the dopamine agonists [pramipexole and ropinirole] still remain as the first-line agents for RLS," says Jack Chen, associate professor, neurology, schools of medicine and pharmacy at Loma Linda University, Loma Linda, Calif.
IMAGING
A gadolinium-based contrast agent, gadobutrol (Gadavist) provides contrast-based imaging of the central nervous system (CNS) for patients-children aged 2 and older and adults-undergoing magnetic resonance imaging. It identifies lesions that interrupt the cell barrier between the brain and blood stream and can detect and visualize abnormal blood supply circulation of the CNS.
One of 6 drugs approved in its class, gadobutrol is more concentrated and requires half the volume of the other drugs. Two clinical trials with 657 patients proved its safety and efficacy.
Gadobutrol carries a boxed warning about the risk of nephrogenic systemic fibrosis, but the drug has been shown to carry less risk than its counterparts.
A contrast agent indicated for striatal dopamine transporter visualization, ioflupane I-123 injection (DaTscan) is used with single-photon emission computed tomography imaging to help detect dopamine transporters in adult patients suspected of having Parkinsonian syndromes.
In 2 phase 3 trials with 284 adult patients with tremor, ioflupane I-123 injection proved to be more effective in detecting the transporters when compared to a "reference clinical diagnosis."
DEPRESSION
Vilazodone hydrochloride (Viibryd) treats major depressive disorder in adults, including feelings of guilt, restlessness, impaired concentration, and suicidal thoughts. The tablets are available in 10-, 20-, and 40-mg doses. Vilazodone hydrochloride tablets carry a boxed warning alerting the possibility of suicidal thoughts among children, adolescents, and young adults; however, the risk of suicidal feelings by adults aged 24 and older and aged 65 and older-the latter on antidepressants-has not increased with the use of vilazodone.
Andes explains that data from clinical trials show that vilazodone is less likely to cause weight gain and negatively impact patients' sexual function-side effects often seen with other antidepressants. "The antidepressant market is large and predominantly generic," she says.
Drugs such as aripiprazole (Abilify) and quetiapine fumarate extended-release (Seroquel XR) tablets, which are used as adjunctive therapy for depressive conditions, also have an opportunity to make a dent in the marketplace.
LICE
Relief for children and their parents comes in the form of spinosad (Natroba) topical suspension 0.9%, which treats head lice infestation for patients aged 4 years and older. It should be applied only to the scalp and hair.
In 2 multicenter, randomized, active-controlled studies, 552 children were treated with spinosad. They received a 10-minute rinse and if live lice were still present after a week, they received a second treatment. Those who were found lice-free with spinosad 14 days after treatment numbered 86% compared to the 44% with permethrin 1%.
"Spinosad may also be a more convenient option because a greater proportion of spinosad-treated patients required only 1 treatment instead of 2, and spinosad use did not require an additional nit combing step," Andes explains.
HEREDITARY ANGIOEDEMA
A new orphan drug-icatibant acetate (Firazyr)-has been approved to treat acute attacks of a rare condition known as hereditary angioedema (HAE). It is caused by low levels or the improper function of a C1 inhibitor-which regulates certain immune system and blood clotting functions-in adults aged 18 years and older. Fewer than 30,000 individuals in the United States have HAE.
It is the first in a new class of drugs treating HAE, but follows 2 drugs approved in 2009 for HAE; C1 esterase inhibitor (human) (Berinert) targets facial and abdominal attacks and ecallantide (Kalbitor) is for patients aged 16 years and older. The disease causes rapid swelling of extremities, limbs, the face, and intestinal and digestive tracts.
Icatibant has an advantage of being self-administered in the abdominal area, enabling patients to treat themselves when they feel an attack coming on.
The drug was tested in 225 patients during 3 controlled clinical trials with open-label extension periods. They received a total of 1076 doses of 30 mg of the drug with median symptom relief in 2 hours compared to 20 hours with a placebo.
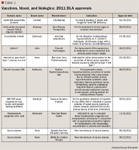
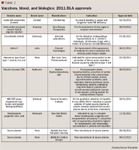
VACCINES, BLOOD, AND BIOLOGICS: BLA APPROVALS
FDA approved the BLA factor XIII concentrate (human) (Corifact) to prevent bleeding in those with the rare genetic defect congenital factor XIII deficiency. This deficiency affects 1 of 3 to 5 persons in the United States. Factor XIII is essential to blood clotting.
Factor XIII concentrate (human) received an orphan-drug designation and fast track approval from FDA. It was approved based on the results of a clinical trial, including children, with the factor XIII deficiency. The condition may cause soft tissue bruising, mucosal bleeding, and fatal intracranial bleeding.
Vogenberg points out that factor XIII concentrate (human), targeting congenital hemophilia, is a purified product with fewer potentially dangerous antibodies and antigens. It is the first FDA-approved product to prevent bleeding in this rare disease.
Apart from factor XIII concentrate (human), the only BLA to receive orphan status is centruroides (scorpion) immune F(ab')2 (equine) injection (Anascorp). Treating clinical signs of scorpion envenomation, it is the first specific treatment for a scorpion sting by Centruroides scorpions in the United States that are most prevalent in Arizona. Their stings can cause local pain immediately with minimal swelling; however, numbness and tingling are often present.
Centruroides (scorpion) immune F(ab')2 (equine) injection is made from the plasma of horses immunized with scorpion venom. Small children are at the highest risk for severe reactions to stings that could be fatal.
The treatment's effectiveness was tested in a randomized, double-blind, placebo-controlled trial of 15 children with neurologic signs of scorpion stings. These signs resolved within 4 hours of treatment in the 8 subjects who received it, but in only 1 of the 7 participants who received the placebo.
The most common side effects are vomiting, fever, rash, nausea, itchiness, headache, runny nose, and muscle pain. In total, safety and efficacy data were collected from 1534 patients in both open-label and blinded studies.
Coccidioides immitis (Spherusol) is a test antigen used to detect a delayed type of hypersensitivity to C. immitis in patients aged 18 to 64 years who have a history of pulmonary coccidioidomy.
Azficel-T (LaViv) improves the appearance of moderate-to-severe nasolabial fold wrinkles (smile or frown lines) in adults. The therapy uses a patient's own skin cells called fibroblasts-used to make collagen-from behind the ears. In turn, the cells are injected into the nasolabial folds. The most common side effects are redness, bruising, swelling, pain, and hemorrhage at the injection site.
Other BLAs approved in 2011 include:
Ms Edlin, a graduate of Stanford University, has been a freelance writer since 1988. Specializing in healthcare, she writes regularly for Managed Healthcare Executive magazine and is a contributor to Drug Topics, Formulary, and Biotechnology Healthcare.
Disclosure Information: The author reports no financial disclosures as related to products discussed in this article.


Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).
Management of antipsychotic medication polypharmacy
June 13th 2013Within our healthcare-driven society, the increase in the identification and diagnosis of mental illnesses has led to a proportional increase in the prescribing of psychotropic medications. The prevalence of mental illnesses and subsequent treatment approaches may employ monotherapy as first-line treatment, but in many cases the use of combination of therapy can occur, leading to polypharmacy.1 Polypharmacy can be defined in several ways but it generally recognized as the use of multiple medications by one patient and the most common definition is the concurrent use of five more medications. The presence of polyharmacy has the potential to contribute to non-compliance, drug-drug interactions, medication errors, adverse events, or poor quality of life.
Medical innovation improves outcomes
June 12th 2013I have been diagnosed with stage 4 cancer of the pancreas, a disease that’s long been considered not just incurable, but almost impossible to treat-a recalcitrant disease that some practitioners feel has given oncology a bad name. I was told my life would be measured in weeks.

