
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Aspirin resistance: a growing concern
Aspirin is the cornerstone of therapy in the treatment and prevention of cardiovascular disease. The potential benefit of aspirin therapy may be significantly reduced in patients with aspirin resistance, creating a clinical and economic burden on the healthcare system. The purpose of this article is to clarify the term "aspirin resistance," describe the proposed mechanisms, review the clinical outcome studies with associated resistance testing, and discuss the potential pharmacologic management of this problem. Literature searches were performed using MEDLINE (January 1966 to January 2006) for review articles on aspirin resistance and antiplatelet activity. Aspirin's primary mechanism of action is to irreversibly inhibit cyclooxygenase-1 (COX-1); however, there are reports of alternative biochemical pathways producing platelet aggregation. The addition of thienopyridines to aspirin should be considered for the management of aspirin-resistant patients. (Formulary. 2006;41:192–201.)
Abstract
Aspirin is the cornerstone of therapy in the treatment and prevention of cardiovascular disease. The potential benefit of aspirin therapy may be significantly reduced in patients with aspirin resistance, creating a clinical and economic burden on the healthcare system. The purpose of this article is to clarify the term "aspirin resistance," describe the proposed mechanisms, review the clinical outcome studies with associated resistance testing, and discuss the potential pharmacologic management of this problem. Literature searches were performed using MEDLINE (January 1966 to January 2006) for review articles on aspirin resistance and antiplatelet activity. Aspirin's primary mechanism of action is to irreversibly inhibit cyclooxygenase-1 (COX-1); however, there are reports of alternative biochemical pathways producing platelet aggregation. The addition of thienopyridines to aspirin should be considered for the management of aspirin-resistant patients. (Formulary. 2006;41:192–201.)
Aspirin is well established in the prevention of cardiovascular disease and is the most widely used antiplatelet therapy today. In addition to preventing cardiovascular events, it is used in patients with vascular grafts, percutaneous angioplasty, acute coronary syndromes, and ischemic strokes. It is estimated that 29 billion tablets are consumed each year by Americans, with the most popular use of aspirin being the prevention of cardiovascular disease.1 However, ex vivo studies have estimated that 5% to 45% of patients taking aspirin are experiencing suboptimal antiplatelet effects.2–6 The impact of aspirin resistance is significant in view of the large population of patients taking aspirin. When considering the direct and indirect costs of treating patients with first-time and recurrent thrombotic events, the problem of aspirin resistance becomes a clinical and economic issue for the healthcare system.

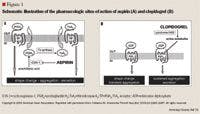
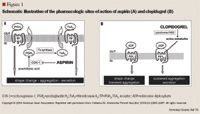
The benefits of aspirin have been studied extensively and its place in therapy has been established through many landmark clinical trials. In the ISIS-2 study, 17,187 patients with suspected myocardial infarction (MI) were randomized to either 160 mg of aspirin daily or placebo. A 23% relative reduction in total vascular mortality was observed with benefits continuing at 15 months follow-up.16
Aspirin has also been shown to be effective in the primary prevention of coronary artery disease (CAD). The US Physician's Health Study17 randomized 22,701 healthy physicians to either 325 mg of aspirin on alternating days or to placebo. The results showed a 44% reduction in the risk of a first MI with the benefits seen primarily in patients aged >50 years.
The Antiplatelet Trialists' Collaboration study18–20 was one of the most comprehensive studies evaluating aspirin in the prevention of cardiovascular events. This meta-analysis included 100,000 patients who were mostly high-risk for vascular disease. Aspirin produced a significant 25% reduction in the triple composite end point of non-fatal MI, non-fatal stroke, or vascular death.
Despite the proven efficacy in clinical studies, there is growing concern regarding patients who continue to experience vascular thrombotic events despite receiving aspirin therapy. Recently, several clinical studies have demonstrated an association between resistance testing and outcomes, shifting this problem to the forefront of current clinical practice.
WHAT IS ASPIRIN RESISTANCE?
Despite the increasing interest in this area, the term "aspirin resistance" has not been clearly defined or uniformly used in research. It has been described in the literature in both clinical and pharmacological terms as: 1) the failure of aspirin to protect against thrombotic complications; 2) the inability of aspirin to cause prolonged bleeding time; 3) the failure of aspirin to inhibit TXA2 production; and 4) the inability of aspirin to prevent platelet function in vitro.21 Investigators have used different indicators to define aspirin resistance depending on the method used to measure platelet aggregation. These methods include: 1) measuring bleeding time; 2) using an optical platelet aggregometer, which measures the amount of light transmitted through a solution of cross-linked platelets; and 3) using an enzyme immunoassay, which measures the amount of a thromboxane metabolite excreted in the urine. Each definition reflects mechanistic failures associated with aspirin and should be considered when evaluating aspirin resistance in a patient.
Experts have also discussed the importance of distinguishing treatment failure from aspirin resistance. Since 60% to 75% of events were not prevented in landmark trials, aspirin therapy should not be expected to prevent all vascular events in clinical practice. The more appropriate term may be "treatment failure" although it is commonly labeled as "aspirin resistance."22 Due to the multiple etiologies and pathways of thrombosis, the failure of aspirin to inhibit TXA2 production and platelet aggregation is only 1 of many causes of treatment failure. To rule out confounders of "true aspirin resistance" such as medication noncompliance,23 laboratory testing should be used to corroborate clinical suspicion if aspirin resistance is speculated to be the cause of a vascular event.
MECHANISMS OF ASPIRIN RESISTANCE
Genetic polymorphism. The response to aspirin may be reduced as a result of a genetic polymorphism of the PlA1 A2 allele for a surface receptor on glycoprotein IIIa. Glycoprotein IIIa is a subunit of the heterodimer glycoprotein (GP) IIb/ IIIa, a fibrinogen receptor involved in the final step leading to platelet aggregation and formation of a thrombus.24 There have been some preliminary data linking the faulty PlA1 A2 allele to increased rates of MI.25 In a separate study, the effects of 300 mg of aspirin were compared between healthy subjects with the homozygous PlA1 A2 allele and those with the polymorphism. Bleeding time was not prolonged in 27% of those with the polymorphism, which may signify an influence of genetics on aspirin-associated platelet function.26
Hypercholesterolemia. Preliminary data suggest that hypercholesterolemia may alter the properties of the platelet cell membrane, resulting in aspirin resistance. This may lead to increased platelet aggregability, enhanced TXA2 production,27,28 and blunted thrombin inhibition by aspirin.29,30
There is evidence that lowering cholesterol in patients with CAD may further reduce thrombin generation in patients receiving aspirin. This effect has been studied clinically. It was concluded that aspirin dosed at 75 mg and 300 mg per day had no effect on thrombin in patients with hypercholesterolemia. After significantly reducing total cholesterol (≤202.8 mg/dL) after 3 months of simvastatin therapy, the total amount of thrombin generation was significantly decreased.29,30 This suggests the effects of aspirin prophylaxis may be inhibited until total cholesterol levels are within the desirable range.31
COX-1 inactivation. The effect of coronary artery bypass graft (CABG) on aspirin resistance has been studied. It has been reported that patients who have undergone CABG procedures may have a reduced response to antiplatelet therapy.20 During in vivo and in vitro studies, platelets displayed delayed inhibition of cyclooxygenase by aspirin, which may be a result of impaired interaction between the aspirin and platelet COX-1.32 However, this postulation remains to be elucidated.
Alternative sources of TXA2. Although TXA2 production is limited by COX-1 inhibition by aspirin, there may be alternative pathways that exist for the production of TXA2. COX-2 is able to convert arachidonic acid to prostaglandin H2, which then can form TXA2 in the platelets.33,34 Vascular endothelial cells and smooth muscle cells have been found to contain alternative COX-2 pathways.35 In addition, TXA2 may be produced from COX-1 in extra platelet sources such as macrophages of vascular endothelial cells.36,37
Oxidative stress. Bioactive prosta-glandin PGF2-like compounds (isoprostanes) are produced from arachidonic acid through lipid peroxidation. These newly discovered compounds are a result of oxygen free radicals acting on cell membranes and LDL particles. The 8-iso-PGF2 alpha causes vasoconstriction and amplifies the response of platelets to agonists.38
Isoprostanes are also associated with cigarette smoking.39,40 Although there are studies that demonstrate the effect of aspirin in preventing platelet aggregation in patients who smoke,41,42 there are also conflicting data revealing the inability of aspirin to prevent platelet aggregation in these patients.43 Further study in this area is required to establish a conclusion.
Exercise-induced platelet activation. Several studies have evaluated the effect of exercise on platelet function in healthy patients as well as patients with CAD receiving aspirin.44–46 The reduced platelet inhibitory effect may be explained through elevations in catecholamines, such as epinephrine and norepinephrine.44 The catecholamine-induced platelet activation is described as an alternative pathway of aspirin resistance that may have a role in acute coronary syndromes. The studies suggest that aspirin may have limited antithrombotic effects during exercise or stress as a result of elevated catecholamine levels.44–46
Increased reactivity of platelets. Aspirin-resistant patients demonstrate increased platelet reactivity to collagen, a physiologically important activating agent, compared to aspirin responders.47
Erythrocytes have also been shown to cause platelet reactivity and thrombosis through stimulation of substances such as adenosine diphosphate (ADP), TXA2, and serotonin (5-HT).48 These factors promote aggregation and contribute to platelet recruitment. Because these factors can be elicited through pathways independent of platelet COX-1, the effects of aspirin on this type of platelet reactivity may be negligible at low doses (50 mg/d).49,50 However, there are data suggesting a 500-mg load every 2 weeks may inhibit erythrocyte-induced stimulation of platelets.50



The clinical significance of this interaction is that it may reduce the cardioprotective effect in patients receiving concomitant administration of aspirin and NSAIDs.52 The clinical effect of the antagonism of platelet inhibition by aspirin was demonstrated in an observational study of secondary prevention of a cardiovascular event. In patients who were receiving low doses of aspirin (<325 mg/d), there was an increased relative risk of all-cause mortality and cardiovascular mortality (RR=1.93; 95% CI, 1.30–2.87 and RR=1.73; 95% CI, 1.05–2.84, respectively) in those who used aspirin plus ibuprofen compared to aspirin alone.52
Further clinical data have supported the hypothesis of a drug interaction between ibuprofen and aspirin. In a post hoc analysis of the Physicians' Health Study, the authors demonstrated that chronic use of NSAIDs, defined by ≥60 days per year, inhibits the clinical benefit of aspirin (325 mg on alternate days) on first MI. There was a >2-fold increased risk of MI in subjects taking chronic NSAIDs and aspirin concomitantly. Intermittent use of NSAIDs (1 to 59 d/y) was not associated with an increased cardiovascular risk in patients receiving aspirin or placebo.57
One study demonstrated that the order of medication administration affected the degree of COX-1 inhibition in patients taking both aspirin and ibuprofen.51 When participants took aspirin 81 mg 2 hours before ibuprofen 400 mg, there was a mean (±SD) degree of inhibition of 99±0.3% in contrast to 53±7% (P< .001) when participants took ibuprofen 2 hours before aspirin. The study found no significant difference in COX-1 inhibition when participants took acetaminophen 1,000 mg or rofecoxib 25 mg 2 hours before aspirin compared to these agents taken 2 hours after aspirin. This provides evidence that the drug interaction between aspirin and NSAIDs similar to ibuprofen can be avoided by administering aspirin at least 2 hours prior to other agents.
Resistance over time. The frequency of aspirin resistance may increase over time. Several studies have demonstrated a progressive reduction in aspirin's ability to inhibit platelet aggregation over long-term treatment.23,58,59 There may be a role for platelet sensitivity testing in aspirin responders who receive chronic therapy.
DOSE OF ASPIRIN
The optimal dose of aspirin should be reconsidered prior to performing platelet function tests in patients who are considered treatment failures. The dose of aspirin should maximize efficacy and minimize toxicity.
Several studies report the antithrombotic effect of aspirin being demonstrated in doses as low as 50 and 100 mg/d.22 Clinical studies have confirmed the efficacy of these lower doses. The risk of MI, death, and stroke were significantly reduced in aspirin doses as low as 75 mg/d; however, acute MI and stroke require minimal effective doses of 160 mg/d.22 Although there are ex vivo data supporting reduced rates of aspirin resistance by increasing the aspirin dosage from 325 to 1,300 mg/d23 , clinical evidence consistently demonstrates that high doses of aspirin are not necessarily more effective than low or moderate doses.20 Moreover, lower doses (300 mg/d) have been shown to cause fewer gastrointestinal side effects compared with higher doses (1,200 mg/d).24
Presenters at the 7th American College of Chest Physicians (ACCP) Conference on Antithrombotic and Thrombolytic Therapy recommend the use of the lowest effective dose of aspirin in order to maximize its efficacy and minimize its toxicity.22 This includes 50 mg daily for transient ischemic attack (TIA) and ischemic stroke; 75 mg daily for men at high cardiovascular risk, with hypertension, stable angina, unstable angina, or severe carotid artery disease; and 160 mg daily for acute myocardial infarction and acute ischemic stroke.
Based on the clinical efficacy and toxicity findings, the dose of aspirin should be prescribed at the lowest recommended dose in both aspirin responders and aspirin non-responders since aspirin resistance may not be improved through escalated doses.
ASPIRIN RESISTANCE IN CLINICAL TRIALS

Other trials have investigated the effect of aspirin resistance on coronary interventions such as PCI and CABG. Chen et al62 demonstrated that aspirin resistance was associated with impaired coronary flow reserve (CFR), a measure of microvascular integrity, in patients undergoing PCI. The study suggested that insufficient aspirin-induced platelet inhibition was a cause of microvascular dysfunction.
Another study by Poston et al63 investigated the clinical outcomes of 225 aspirin-resistant patients receiving CABG surgery. The incidence of aspirin resistance was 30% in this study population, and the authors found that graft thrombosis was associated with aspirin resistance (P<.04). The incidence of aspirin resistance was significantly higher in patients with early graft failure compared to those with patient grafts. Thus these studies further establish the relationship between aspirin resistance and adverse clinical outcomes of coronary interventions.
Several clinical trials have also examined a possible relationship between aspirin resistance and clopidogrel resistance as well as the effect of this relationship on clinical end points. Eikelboom et al64 found an inverse correlation between the antiplatelet effects of clopidogrel and aspirin in 36 patients with symptomatic peripheral arterial disease. Patients experiencing the greatest levels of platelet aggregation inhibition with aspirin had the least inhibition of platelet aggregation with clopidogrel (mean reduction 2.8%; 95% CI, 0.8%-6.3%). Similarly, patients with the least inhibition by aspirin had the greatest inhibition of platelet aggregation with clopidogrel (mean reduction 12.6%; 95% CI, 4.5%–20.8%). The study suggests that adding clopidogrel to aspirin may have the greatest clinical benefit in patients whose platelets are least inhibited by aspirin. Since this study used surrogate endpoints, confirmation with studies using clinical outcomes may show that aspirin resistance may be overcome with the addition of clopidogrel.
However, a small study by Lev et al65 found the opposite relationship between aspirin resistance and clopidogrel resistance. The study assessed aspirin and clopidogrel resistance in 150 patients undergoing elective percutaneous coronary intervention (PCI) . It found that a significantly greater percentage of aspirin-resistant patients were also resistant to clopidogrel compared to aspirin-sensitive patients. However, the incidence of aspirin resistance was low (19 patients, 12.7%) and the study was likely not adequately powered for conclusive evidence.
MANAGEMENT OF PATIENTS WITH ASPIRIN RESISTANCE
The clinical management of patients with aspirin resistance is still under investigation. Theoretically, the addition of thienopyridines such as ticlopidine and clopidogrel may seem to be an ideal method of overcoming aspirin resistance since they inhibit platelet aggregation independently of aspirin. These agents irreversibly inhibit ADP from binding at the platelet receptors, preventing fibrinogen binding, platelet adhesion, and platelet aggregation (Figure 1). There are data suggesting enhanced platelet inhibition with the combination of both classes of drugs.66–69 Dual antiplatelet therapy with ticlopidine plus aspirin has been determined to be superior to aspirin alone in coronary stenting.66 However, ticlopidine has been largely replaced by clopidogrel in clinical practice due to ticlopidine-associated neutropenia and bone marrow suppression.70 Furthermore, large clinical trials have demonstrated significant benefits associated with the combined use of aspirin and clopidogrel compared to aspirin alone in patients with stroke and acute coronary syndromes.71–73 There are no data available on the number of aspirin-resistant patients in these studies, or if a clinical benefit was associated with adding clopidogrel to aspirin. Despite the risk of major and minor bleeding complications associated with dual therapy, there may be a role for this combination in aspirin-resistant patients.
LABORATORY TESTING OF ASPIRIN AGGREGATION
The clinical studies have demonstrated an association between major events and aspirin resistance based on platelet aggregation tests; however, the limitations in each study reduce the clinical application of the information. Several methods of platelet function tests have been developed to measure platelet aggregation and aspirin resistance. The optical aggregometry in citrated platelet-rich plasma is considered to be the "gold standard" and possibly the most common test.74 Two point-of-care testing modalities, the PFA-100 (Dade-Behring, Deerfield, Ill) and the VerifyNow Aspirin Assay (Accumetrics, San Diego, Calif), are available and may be more convenient for routine clinical practice. These products have been shown to identify patients who are aspirin nonresponders, but the correlation to clinical events has yet to be established. Since the clinical consequences of these tests are unclear, further research in this area is required to elucidate a cause and effect between aspirin resistance and clinical outcomes.
COST IMPLICATIONS
Therapeutic failure due to aspirin resistance can have a major impact on the cost of treating patients with coronary heart disease and stroke. The American Heart Association (AHA) estimates that $112 billion per year is spent on direct costs, including hospital, nursing home, physician, and drug costs, on these diseases. The cost on society adds another $88 billion per year in lost productivity and loss of future earnings.75 AHA also estimates that 2.4 million procedures, including angioplasty, PCI, and cardiac revascularization, are performed yearly. Since aspirin resistance prevalence is estimated to be between 5% and 45%, the cost of treatment and the number of interventions can be significantly reduced if patients can be identified with validated laboratory tests and treated appropriately according to their resistance to aspirin.
CONCLUSIONS
Aspirin resistance continues to be a problem, particularly in patients with cardiovascular disease and ischemic events. A clear definition of aspirin resistance remains to be established. Dual therapy with aspirin and clopidogrel may be an alternative in aspirin-resistant patients who have been identified by optical platelet aggregability testing or elevated urinary concentrations of 11-dehydro- thromboxane B2; however, more information is required before a specific test can be recommended for routine use. Although point-of-care testing allows for accessible, rapid results, the appropriate clinical response to these results is still to be determined. Platelet function tests are not recommended for routine monitoring at this time and more prospective studies on clinical outcomes must be done. Once this information is obtained, patients who experience treatment failure on aspirin therapy should be tested for platelet aggregation if there are no other identifiable causes for treatment failure. Based on the current understanding of aspirin resistance, further studies are needed to clarify the relationship between platelet aggregation, resistance testing, pharmacologic management, and clinical outcomes.
Dr Chow is an assistant professor of pharmacy practice, cardiology, Western University of Health Sciences, College of Pharmacy, Pomona, Calif. She can be reached at schow@westernu.edu
. Dr Cheung is an assistant professor of pharmacy practice, Loma Linda University, School of Pharmacy, Loma Linda, Calif.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. Bayer Pharmaceuticals. Facts about aspirin. Available at: http:// http://www.wonderdrug.com/press/factsheets/aspirin_fact_sheet.pdf. Accessed March 23, 2006.
2. Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88:230–235.
3. Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke. 1993;24:345–350.
4. Pappas JM, Westengard JC, Bull BS. Population variability in the effect of aspirin on platelet function. Implications for clinical trials and therapy. Arch Pathol Lab Med. 1994;118:801–804.
5. Grotemeyer KH. Effects of acetylsalicylic acid in stroke patients. Evidence of nonresponders in a subpopulation of treated patients. Thromb Res. 1991;63:587–593.
6. Valles J, Santos MT, Aznar J, et al. Erythrocyte promotion of platelet reactivity decreases the effectiveness of aspirin as an antithrombotic therapeutic modality: the effect of low-dose aspirin is less than optimal in patients with vascular disease due to prothrombotic effects of erythrocytes on platelet reactivity. Circulation. 1998;97: 350–355.
7. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets: I. Acetylation of a particulate fraction protein. J Clin Invest. 1975;56:624–632.
8. Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci USA. 1975;72:3073–3076.
9. Burch JW, Stanford N, Majerus PW. Inhibition of platelet prostaglandin synthetase by oral aspirin. J Clin Invest. 1978;61:314–319.
10. Majerus PW. Arachidonate metabolism in vascular disorders. J Clin Invest. 1983;72:1521–1525.
11. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271: 33,157–33,160.
12. Cattaneo M, D'Angelo A, Canciani MT, et al. Effect of oral aspirin on plasma levels of vitamin K-dependent clotting factors-studies in healthy volunteers (Letter). Thromb Haemost. 1988;59:540.
13. Buczko W, Mogielnicki A, Kramkowski K, Chabielska E. Aspirin and the fibrinolytic response. Thromb Res. 2003;110:331–334.
14. Husain S, Andrews NP, Mulcahy D, Panza JA, Quyyumi AA. Aspirin improves endothelial dysfunction in atherosclerosis. Circulation. 1998;97:716–720.
15. Kharbanda RK, Walton B, Allen M, et al. Prevention of inflammation-induced endothelial dysfunction: a novel vasculo-protective action of aspirin. Circulation. 2002;105:2,600–2,604.
16. ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet. 1988; 2:349–360.
17. Steering Committee of the Physicians' Health Study Research Group. Final report on the aspirin component of the ongoing Physicians' Health Study. N Engl J Med. 1989;321:129–135.
18. Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy-I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ. 1994;308:81–106.
19. Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy-II: Maintenance of vascular graft or arterial patency by antiplatelet therapy. BMJ. 1994;308:159–168.
20. Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy-III: Reduction in venous thrombosis and pulmonary embolism by antiplatelet prophylaxis among surgical and medical patients. BMJ. 1994;308:235–246.
21. Patrono C. Aspirin resistance: definition, mechanisms and clinical read-outs. J Thromb Haemost. 2003;1:1710–1713.
22. Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004; 126(Suppl 3):234S–264S.
23. Schwartz KA, Schwartz DE, Ghosheh K, Reeves MJ, Barber K, DeFranco A. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol. 2005;95:973–975.
24. Rozalski M, Boncler M, Luzak B, Watala C. Genetic factors underlying differential blood platelet sensitivity to inhibitors. Pharmacol Rep. January–February 2005;57:1–13.
25. Weiss EJ, Bray PF, Tayback M, et al. A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med. 1996;334:1090–1094.
26. Szczeklik A, Undas A, Sanak M, Frolow M, Wergrzyn W. Relationship between bleeding time, aspirin, and the PIA1/A2 polymorphism of platelet glycoprotein IIIa. Br J Haemotol. 2000; 110:965–967.
27. Tremoli E, Colli S, Maderna P, Baldassarre D, Di Minno G. Hypercholesterolemia and platelets. Semin Thromb Hemost. 1993;19:115–121.
28. Davi G, Averna M, Catalano I, et al. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation. 1992;85:1792–1798.
29. Szczeklik A, Musial J, Undas A, et al. Inhibition of thrombin generation by simvastatin and lack of additive effects of aspirin in patients with marked hypercholesterolemia. J Am Coll Cardiol. 1999;33:1286–1293.
30. Musial J, Undas A, Undas R, Brozek J, Szczeklik A. Treatment with simvastatin and low-dose aspirin depresses thrombin generation in patients with coronary heart disease and borderline-high cholesterol levels. Thromb Haemost. 2001;85: 221–225.
31. Szczeklik A, Musial J, Undas A, et al. Aspirin and thrombinogenesis. Thromb Res. 2003;110: 345–347.
32. Zimmermann N, Wenk A, Kim U, et al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery. Circulation. 2003;108:542–547.
33. Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–845.
34. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120.
35. Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–126.
36. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105: 1650–1655.
37. Halushka MK, Halushka PV. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2? Circulation. 2002;105:1620–1622.
38. Cipollone F, Ciabattoni G, Patrignani P, et al. Oxidant stress and aspirin-insensitive thromboxane biosynthesis in severe unstable angina. Circulation. 2000;102:1007–1013.
39. Morrow JD, Frei B, Longmire AW, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332: 1198–1203.
40. Reilly M, Delanty N, Lawson JA, FitzGerald GA. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation. 1996;94:19–25.
41. Davis JW, Davis RF. Prevention of cigarette smoking-induced platelet aggregate formation by aspirin. Arch Intern Med. 1981;141:206–207.
42. Davis JW, Davis RF, Hassanein KM. In healthy habitual smokers acetylsalicylic acid abolishes the effects of tobacco smoke on the platelet aggregate ratio. Can Med Assoc J. 1982;126:637–639.
43. Davis JW, Hartman CR, Lewis HD Jr, et al. Cigarette smoking-induced enhancement of platelet function: lack of prevention by aspirin in men with coronary artery disease. J Lab Clin Med. 1985;105:479–483.
44. Larsson PT, Wallen NH, Hjemdahl P. Norepinephrine-induced human platelet activation in vivo is only partly counteracted by aspirin. Circulation. 1994;89:1951–1957.
45. Christiaens L, Macchi L, Herpin D, et al. Resistance to aspirin in vitro at rest and during exercise in patients with angiographically proven coronary artery disease. Thromb Res. 2002;108: 115–119.
46. Hurlen M, Seljeflot I, Arnesen H. Increased platelet aggregability during exercise in patients with previous myocardial infarction. Lack of inhibition by aspirin. Thromb Res. 2000;99: 487–494.
47. Kawasaki T, Ozeki Y, Igawa T, Kambayashi J. Increased platelet sensitivity to collagen in individuals resistant to low-dose aspirin. Stroke. 2000;31:591–595.
48. Marcus AJ. Platelets and their disorders. In: Ratnoff OD, Forbes CD, eds. Disorders of Hemostasis. Philadelphia, PA: Grune & Stratton;1990;75.
49. Valles J, Santos MT, Aznar J, et al. Erythrocytes metabolically enhance collagen-induced platelet responsiveness via increased thromboxane production, adenosine diphosphate release, and recruitment. Blood. 1991;78:154–162.
50. Santos MT, Valles J, Aznar J, Marcus AJ, Broekman MJ, Safier LB. Prothrombotic effects of erythrocytes on platelet reactivity. Circulation. 1997;95:63–68.
51. Catella-Lawson F, Reilly M, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345: 1809–1817.
52. MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573–574.
53. Livio M, Del Maschio A, Cerletti C, de Gaetano G. Indomethacin prevents the long-lasting inhibitory effect of aspirin on human platelet cyclo-oxygenase activity. Prostaglandins. 1982;23: 787–796.
54. Rao GH, Johnson GG, Reddy KR, White JG. Ibuprofen protects platelet cyclooxygenase from irreversible inhibition by aspirin. Arteriosclerosis. 1983;3:383–388.
55. Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol. 1995;2:637–643.
56. Loll PJ, Picot D, Ekabo O, Garavito RM. Synthesis and use of iodinated nonsteroidal antiinflammatory drug analogs as crystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site. Biochemistry. 1996;35:7330–7340.
57. Kurth T, Glynn RJ, Walker AM, et al. Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal antiinflammatory drugs. Circulation. 2003;108:1191–1195.
58. Pulcinelli FM, Pignatelli P, Celestini A, Riondino S, Gazzaniga PP, Violi F. Inhibition of platelet aggregation by aspirin progressively decreases in long-term treated patients. J Am Coll Cardiol. 2004;43:979–984.
59. Andersen K, Hurlen M, Arnesen H, Seljeflot I. Aspirin non-responsiveness as measured by PFA-100 in patients with coronary artery disease. Thromb Res. 2002;108:37–42.
60. Gum PA, Kottke-Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–965.
61. Mueller MR, Salat A, Stangl P, et al. Variable platelet response to low-dose ASA and the risk of limb deterioration in patients submitted to peripheral arterial angioplasty. Thromb Haemost. 1997;78:1003–1007.
62. Chen WH, Lee PY, Ng W, et al. Relation of aspirin resistance to coronary flow reserve in patients undergoing elective percutaneous coronary intervention. Am J Cardiol. 2005;96: 760–763.
63. Poston RS, Gu J, Brown JM, et al. Endothelial injury and acquired aspirin resistance as promoters of regional thrombin formation and early vein graft failure after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2006;131:122–130.
64. Eikelboom JW, Hankey GJ, Thom J, et al. Enhanced antiplatelet effect of clopidogrel in patients whose platelets are least inhibited by aspirin: a randomized crossover trial. J Thromb Haemost. 2005;3:2649–2655.
65. Lev EI, Patel RT, Maresh KJ, et al. Aspirin and clopidogrel drug response in patients undergoing percutaneous coronary intervention: the role of dual drug resistance. J Am Coll Cardiol. 2006;47:27–33.
66. Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting, Stent Anticoagulation Restenosis Study Investigators. N Engl J Med. 1998;339:1665–1671.
67. Herbert JM, Dol F, Bernat A, Falotico R, Lale A, Savi P. The antiaggregating and antithrombotic activity of clopidogrel is potentiated by aspirin in several experimental models in the rabbit. Thromb Haemost. 1998;80:512–518.
68. Bossavy JP, Thalamas C, Sagnard L, et al. A double-blind randomized comparison of combined aspirin and ticlopidine therapy versus aspirin or ticlopidine alone on experimental arterial thrombogenesis in humans. Blood. 1998;92:1518–1525.
69. Moshfegh K, Redondo M, Julmy F, et al. Antiplatelet effects of clopidogrel compared with aspirin after myocardial infarction: enhanced inhibitory effects of combination therapy. J Am Coll Cardiol. 2000;36:699–705.
70. Cazenave JP, Gachet C. Pharmacology of ticlopidine and clopidogrel. In: Gresele P, Page C, Fuster V, Vermylen J, eds. Platelets in Thrombocyte and non-Thrombotic Disorders. Cambridge: Cambridge University Press; 2002:929–939.
71. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; for the Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494–502.
72. Steinhubl SR, Berger PB, Mann JT III, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;288:2411–2420.
73. Mehta SR, Yusuf S, Peters RJ, et al; for the Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527–533.
74. Haubelt H, Anders C, Hellstern P. Can platelet function tests predict the clinical efficacy of aspirin? Semin Thromb Hemost. 2005;31:404–410.
75. Thom T, Haase N, Rosamond W, et al; for the The American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2006 Update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151.
76. Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24:1980–1987.

FDA Approves Combination Therapy for Pulmonary Arterial Hypertension
March 25th 2024J&J’s Opsynvi is single-tablet combination of macitentan, an endothelin receptor antagonist, and tadalafil, a PDE5 inhibitor. It will be priced on parity with Opsumit, which is also a J&J product to treat patients with PAH.




FDA Issues Complete Response Letter for Onpattro in Heart Failure Indication
October 9th 2023Alnylam Pharmaceuticals will no longer pursue this indication of Onpattro and will instead on focus on a label expansion for Amvuttra, which is in phase 3 development to treat patients with cardiomyopathy of ATTR amyloidosis.


