
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Biosimilars and the Patient Protection and Affordable Care Act: Where do we stand?
Among the long-awaited provisions in PPACA are the additions to the Public Health Service Act that allow for the introduction of biosimilar medications to the marketplace.



Key Points
Abstract
The recent passage of the Patient Protection and Affordable Care Act (PPACA) will generate unprecedented changes to the healthcare system of the United States. Among the long-awaited provisions in PPACA are the additions to the Public Health Service Act that allow for the introduction of biosimilar medications to the marketplace. The advent of these medications is projected to save billions of healthcare dollars annually. In the near future, FDA will release its final guidance on application requirements, patent protection, and user fees. Other considerations that will be of interest are immunogenicity and prescriber acceptance of biosimilars as key biologic drug patents expire. (Formulary. 2011; 46:474–485.)
In 1982 Eli Lilly launched Humulin insulin, the first genetically engineered pharmaceutical product available. By the end of 1993 sales had reached approximately $560 million.1 Those figures represent the beginning of a huge growth trend in biologic drugs, with the top 16 biologic classes product sales at $107.69 billion in 2010.2
Currently, biologic drugs are regulated under Public Health Service (PHS) Act section 351, which provides a definition for biologic drugs as a "virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product,…applicable to the prevention, treatment, or cure of a disease or condition of human beings."3
PPACA AND BIOSIMILARS
The 2010 historic Patient Protection and Affordable Care Act (PPACA), also known as the healthcare reform bill, will affect the healthcare sector in unprecedented ways. One of the provisions in PPACA will reshape the regulatory pathway for biologic medications by allowing generic versions to be produced.4 Because it would be difficult to produce a copy of a biologic protein, these medications are known as biosimilars or "follow-on biologics."
Provisions of PPACA that impact life sciences or biotech companies are sections 3139 and 7001 to 7003, also known as the Biologics Price Competition and Innovation Act of 2009 (BPCIA).4 The BPCIA adds sections k, 1, and m to the PHS Act section 351 to allow licensure of a biologic drug shown to be biosimilar to a branded reference product.4
The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman amendments, created an accelerated pathway for approval of generic small molecule drugs.5 The Federal Food, Drug and Cosmetic (FDC) Act 505(j) states that a drug must show safety and effectiveness and bioequivalence.6 It would be difficult to achieve this sort of threshold for biologics due to the challenges presented by manufacturing, immunogenicity, and surveillance.3 Most biologic drugs are regulated under the PHS Act and not under the Federal FDC Act, which governs conventional medicines. The recent additions of sections k, 1, and m to the PHS Act will provide similar guidance.4
BIOSIMILAR APPLICATION REQUIREMENTS
In particular an application submitted under the new sections of the PHS Act section 351 should include the following: 1) the biological product that is the subject of the applications is "biosimilar" to a single reference product; 2) the biological product and reference product use same mechanism(s) of action for the condition(s) of use prescribed, recommended, or suggested in proposed labeling, but only to extent the mechanism(s) of action are known for the reference product; 3) the condition(s) of use prescribed, recommended, or suggested in the proposed labeling for the biological product have been previously approved for the reference product; 4) the biological product has same route of administration, dosage form, and strength as the reference product; and 5) the facility in which the biological product is manufactured, processed, packed, or held meets standards designed to assure the biological product continues to be safe, pure, and potent.7
Manufacturers of biosimilar medications may also seek a determination of interchangeability, either at the time of the original application or as a later supplement. FDA may grant interchangeability if the information submitted in the application shows both that the biological product is biosimilar to the reference product and the biological product can be expected to produce the same clinical result as the reference product in any given patient.7 In addition, FDA must determine the safety risks of switching between the 2 products, finding that switching between products produces no greater risk than that for continuing to use the reference product. Interchangeable products may be substituted for the reference drug and are eligible for 1 year of market exclusivity. Only the first product determined to be interchangeable will be granted exclusivity with respect to interchangeability.7
CHALLENGES TO IMPLEMENTATION
Of interest to payers is the amendment to section 351(i) of the PHS Act that allows products judged to be interchangeable to be substituted for the reference product without the approval of the prescriber.4 According to a survey conducted in 2010, healthcare executives believe that biosimilars will be available at approximately 40% discount from branded biologic drug products.8 This provision provides patients and pharmacists greater autonomy by allowing a substitution of an interchangeable biologic for the reference product without prescriber intervention.
It seems inevitable that patent disputes will arise between the manufacturer of the reference product and the biosimilar applicant. In contrast to the Hatch-Waxman amendments, however, the sponsor of the reference product and the biosimilar applicant share information concerning the manufacturing process of the biosimilar.4 Within 20 days of notification from FDA of acceptance for review, the biosimilar applicant sends a copy of the application and information relating to their manufacturing process to the reference sponsor. This provision allows the reference product sponsor to determine if the biosimilar applicant product would infringe on any patents owned or licensed by the reference product sponsor. The reference product sponsor must provide a list of possible patent infringements to the biosimilar applicant within 60 days of receiving their material. The biosimilar applicant then has 60 days to provide a list of its own patents to the reference product sponsor, that the applicant reasonably believes that the reference drug sponsor could claim infringement. After the lists of possible infringements have changed hands, the parties will engage in good faith negotiations and the determination of which patents are to be litigated will be made. As part of an expedited litigation procedure, if the parties cannot agree within 15 days, they must exchange lists of patents that each party believes should be asserted, and the biosimilar applicant may choose as many as they desire, while the reference product sponsor may choose at least 1 but no more than the biosimilar applicant.4 It is unclear at this time how the newly enacted Patent Reform Act of 2011 will affect biosimilar applications. The Patent Reform Act encompasses several provisions including sections dealing with a first-to-file system, as is used in most other countries, rather than the current first-to-invent system.9
Not unlike the other user fee system for small molecule drugs, applications submitted under section 351(k) will use the same user fee system for biosimilar applications. Costs of reviewing biosimilar applications will be audited, and if necessary, user fees will be altered to account for differences in costs. Starting in October 2010, FDA must develop performance goals for biosimilar application review in the years 2012 to 2017.4
Integrating the new regulatory framework to the science of producing biosimilar medications will pose numerous challenges. The first issue at hand is to establish scientific criteria for defining similarity. In regard to the complex protein structures of biologics, "the process is the product." Whereas small molecule drugs are relatively simple to reproduce, protein-based drugs can exhibit inherent variability. Although smaller proteins such as insulin and growth hormone may be relatively simple to reproduce due to their well-known properties, many protein-based therapies are hundreds to many thousand times larger, and exhibit a highly complex 3-dimensional structure. Alterations to the structure, including isomers and glycosylation, may lead to a product's heterogeneity.10
THE EUROPEAN EXPERIENCE
The European Medicines Agency (EMA) published general guidelines on biosimilars in 2005.11 It approved its first biosimilar product, the growth hormone Omnitrope (somatropin [rDNA origin] injection), in 2006.12 EMA guidelines are "class specific," meaning products are grouped together, such as monoclonal antibodies. The guidelines outline study design activities, biomarkers, and clinical end points that could produce potential differences from the reference products. Differences in the impurity profiles are considered as well on a case-by-case basis.11 Since the end of 2008, FDA has collaborated with the EMA on their experience with biosimilar products, and in 2009, FDA experts attended the EMA workshop on biosimilar monoclonal antibodies. FDA favors a "totality of the evidence" approach to making regulatory decisions, and it seems likely that this approach will be adopted for biosimilars. This approach generally combines animal, human, and structural studies to highlight potential differences in products. A recent EMA draft guideline on biosimilar monoclonal antibodies outlines the concepts relevant to design of biosimilar studies and will be an important consideration in designing a US policy.13 The hope is to explore further enhanced collaboration in the area of biosimilars.12
One of the inherent difficulties with any protein-based therapy is the risk of immunogenicity, or the ability of a product to provoke an immune response. Even the slightest alteration in protein structure may result in reduced efficacy, or immunogenicity, and as a consequence products can vary even from batch to batch. Although the primary sequence of amino acids may be identical for a follow-on product, the secondary and tertiary structures may differ.12 An often-cited example of an immunogenic response is the case of Johnson & Johnson's European-marketed product Eprex (epoetin alfa). Although Eprex, an erythropoietin, is not a biosimilar, this example emphasizes some of the unpredictability not only of protein manufacturing but also of the human immune system. In Europe in 2001, approximately 175 cases of pure red cell aplasia, a potentially serious type of anemia, occurred. Critics were quick to point out the dangers of manufacturing changes and storage conditions causing the problem. According to Johnson & Johnson, who have studied this problem for years, it stemmed from polysorbate 80 that caused compounds to leach from rubber stoppers in some of the pre-loaded Eprex syringes, and may have caused an enhanced immune response to the protein.14,15
SURVEILLANCE
Given that various factors can cause immunogenic reactions, it is difficult to predict whether a change in protein structure will trigger an immune response. The vast majority of protein-based drugs, with the exception of colony-stimulating factors, trigger antibody production, usually at a subclinical level.15 The main factors in protein-based drugs that cause immunogenic reactions are impurities and aggregates, impurities triggering T-helper cells and aggregates leading to antibody induction.10 Characteristics of the products themselves may also influence antibody induction. Agents that are immunostimulatory in nature might display a higher incidence of antibody induction than immunosuppressive agents. Patient characteristics, such as genetic background, concomitant medications, and the disease itself are also factors that contribute to immunogenic response.10 Additional considerations are duration of treatment, dosage schedule, and route of administration.
Preclinical data are of limited use in predicting immunogenicity in humans, and can be complicated by the complex pharmacokinetic and pharmacodynamic relationships of biologic drugs. Species-specific action and immunogenic properties of animal-derived proteins are of limited value as these apply to humans. A study of safety-related regulatory actions for biologicals that appeared in the Journal of the American Medical Association in 2008 highlights both the nature of adverse effects and the timing of regulatory action.16 During the period of review for the study, which lasted from January 1995 to June 2007, 46 safety-related regulatory actions defined as dear healthcare professional letters (DHPLs) were issued for 30 biologicals in the United States. Adverse reactions and warnings issued for the various biologicals included:
Of further interest is that the length of time from drug approval date to time of DHPLs varied from 0.2 years for antibody production events to 10.5 years for some malignancies. Black box warnings issued in the United States showed a similar pattern with respect to the time until a warning appeared. Although the study was conducted in both the United States and the European Union, the data reflect the uncertainty surrounding safety issues as these apply to biologic drugs. The study demonstrated that of the 174 biologicals included 14% had a probability to require safety-related regulatory action within the first 3 years of marketing approval and 29% within 10 years.16
PAYER ACCEPTANCE
Much speculation has surrounded the impact of biosimilars on costs related to biologic drugs. Originally it was believed that biosimilar medications would offer savings in proportion to those generated by generic small molecule drugs. Realistic estimates of savings are now believed to be approximately 30% lower than the reference drug.17,18 Much of the proposed language in the final guidance will exert an influence on the approval process and ultimately on drug pricing. If payers proactively make changes to their formulary management techniques to allow easier adoption of biosimilar medications, prescribers will likely accept the changes. Tier placement, copays, and prior authorization will serve as incentives for the use of biosimilar medications.
Estimates of national health expenditures under the Centers for Medicare and Medicaid Services were expected to increase by a modest 3.9% in 2010, but increased by 5.2% in 2011.19
Biologic drugs represent a large portion of spending in Medicare Part B, especially erythropoietin agents used in the treatment of end-stage renal disease. In 2004 alone, approximately $1.8 billion were spent on erythropoietin stimulating agents for patients enrolled in Medicare Part B.20 At the estimated savings figure of 30%, use of biosimilar medications would have saved approximately $550 million. The payment provision for biosimilar medications under PPACA for Medicare Part B states that "certain Part B biosimilar generic drugs will be paid at the sum of the average sales price methodology using the National Drug Codes assigned to such products and 6% of the amount determined for the reference biological product."21 This will also apply to biosimilar medications purchased through Medicare Part D.
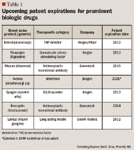
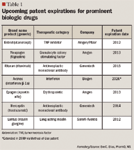
UPCOMING US PATENT EXPIRATIONS
Estimates of worldwide sales of protein-based drug therapies for 2009 are more than $91 billion according to the 2011 Medco Drug Trend Report, and should exceed $150 billion by the end of 2015.22
Several factors will influence the rapid spending growth in this category, including formulary management techniques and the patent expiration of many biologic drugs. In the United States, by the end of 2015, 32 biologics will potentially lose patent protection. These 32 drugs represented approximately $51 billion in sales in 2009 alone.22
Although FDA states that they are ready to review biosimilar applications, it is unknown how long that process may take after it is begun. It seems likely that the first product applications will come under considerable scrutiny that could potentially slow the application and consequently the approval process. Although no biosimilar drugs have been approved in the United States, many are in development.
Of note, the Teva biosimilar filgrastim, which was submitted as a Biologics License Application (BLA) received a complete response letter with approval from FDA in early 2010.23

WHERE ARE WE NOW?
In late September, FDA appeared to be near releasing guidance on biosimilars and will likely have this information available prior to the end of the year. Of interest in the upcoming guidance will be the terminology used to describe the biosimilar products. Misinterpretation of the term biosimilar has led to potentially negative perceptions and "impaired acceptance" of these medications by prescribers and patients.25 A recent article from Nature Biotechnology outlined the problems arising from imprecise terminology. Members of the Biosimilar Medicinal Products Working Party (BMWP) at EMA have created a proposal for more precise terminology for biologic products, and include the terms "biosimilar," "noninnovator biologic," "second-generation biologic," or "biobetter."25 Differences in the definitions as well as clinical implications are important considerations as prescribers utilize this information to safely and efficaciously treat patients. Other stakeholders such as patients, advocacy networks, and managed care organizations will also have an interest in the upcoming guidance from FDA. Hopefully, the guidance will be flexible enough to allow for the evolution of new technology and economic incentives, while maintaining patient safety.
CONCLUSION
The PPACA has provided an approval pathway for biosimilar medications that will balance scientific and regulatory issues while allowing competition. FDA will continue to work closely with the EMA to increase collaboration in the area of biosimilars. In addition to comparing guidelines and regulations with the EMA, FDA should continue to explore opportunities for prescriber and patient education, and the unique challenges to pharmacovigilance that biosimilar medications present.
Dr Elias is director of drug information and clinical assistant professor, University of Missouri Kansas City School of Pharmacy, Columbia Satellite Campus, Columbia.
Disclosure Information: The author reports no financial disclosures as related to products discussed in this article.
REFERENCES
1. Lee KB Jr, Burrill GS. Biotech 95: Reform, Restructure, Renewal. Palo Alto, CA: Ernst & Young; 1994.
2. Top 30 Biologics 2010. R&D Pipeline News. March 3, 2011. Available at http://www.pipelinereview.com/free-downloads/TOP_30_Biologics_2010_RDPN_Special_Mar_2011.pdf. Accessed October 27, 2011.
3. FDA. U.S. Food and Drug Administration. Frequently asked questions about therapeutic biological products. Available at http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm113522.htm. Accessed October 25, 2011.
4. Kingham RF, Lietzan E, Elikan J, et al; Covington & Burling, LLP. Health care reform: biosimilars licensing and reimbursement. April 2010. Available at http://www.cov.com/files/Publication/e0c09313-1b79-4718-a211-dcafa0682a9c/Presentation/PublicationAttachment/f568ca84-aa98-4315-bb20-df1064c6a014/Health%20Care%20Reform%20-%20%20Biosimilars%20Licensing%20And%20Reimbursement.pdf. Accessed October 25, 2011.
5. FDA. U.S. Food and Drug Administration. Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Amendments). August 1, 2003. Available at http://www.fda.gov/newsevents/testimony/ucm115033.htm. Accessed October 26, 2011.
6. FDA. U.S. Food and Drug Administration. Regulatory Information. FD&C Act Chapter V: Drugs and Devices. SEC. 505 [21 USC 355] New Drugs. Available at http://www.fda.gov/regulatoryinformation/legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/ucm108125.htm. Accessed October 26, 2011.
7. FDA. U.S. Food and Drug Administration. Biologics Price Competition and Innovation Act, 2009. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf. Accessed October 25, 2011.
8. Nash DB, Toscani M, Vogenberg FR. The Biologic Finance and Access Council 2nd Annual Biologics Healthcare Survey: views from key healthcare stakeholders. Biotechnology Healthcare. Summer 2011:19–21.
9. Goldman D. CNNMoney. Sweeping patent changes poised to become law. September 9, 2011. Available at http://money.cnn.com/2011/09/09/technology/patent_reform_bill/index.htm. Accessed October 26, 2011.
10. Schellekens H. When biotech proteins go off-patent. Trends Biotechnol. 2004;22(8):406–410.
11. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. 22 February 2006. EMEA/CHMP/BWP/49348/2005. Available at http://www.ema.europa.eu/ . Accessed October 26, 2011.
12. Kozlowski S, Woodcock J, Midthun K, Sherman RB. Developing the nation's biosimilars program. N Engl J Med. 2011;365(5):385–388.
13. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies. Draft. 18 November 2010. EMA/CHMP/BMWP/403543/2010. Available at http://www.ema.europa.eu/. Accessed October 26, 2011.
14. Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int. 2005;67(6):2346–2353.
15. Ledford H. The same but different. Nature. 2007;449(20):274–276.
16. Giezen TJ, Mantel-Teeuwisse AK, Straus SM, et al. Safety-related regulatory actions for biologicals approved in the United States and the European Union. JAMA. 2008;300(16):1887–1896.
17. Wroblewski MS, Jex EA, Munck SD, et al. Emerging Health Care Issues: Follow-on Biologic Drug Competition. Federal Trade Commission Report. June 2009. Available at http://www.ftc.gov/os/2009/06/P083901biologicsreport.pdf. Accessed October 27, 2011.
18. Shapiro RJ, Singh K, Mukim M. The Potential American Market for Generic Biological Treatments and the Associated Cost Savings. Insmed Inc; February 2008. Available at http://www.insmed.com/pdf/Biogeneric_Savings.pdf. Accessed October 27, 2011.
19. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures-2011. Am J Health-Syst Pharm. 2011;68:921–932.
20. Engel & Novitt, LLP. Potential savings that might be realized by the Medicare program from enactment of legislation such as the Access to Life-Saving Medicine Act (H.R. 6257/D.4016) that establishes a new cBLA pathway for follow-on biologics. A report to Pharmaceutical Care Management Association (PCMA) based upon a preliminary assessment of available data. January 2, 2007.
21. The Boards of Trustees Federal Hospital Insurance and Federal Supplementary Medical Insurance Trust Funds. The 2010 Annual Report of the Boards of Trustees of the Federal Hospital Insurance and Federal Supplementary Medical Insurance Trust Funds. Washington, DC: August 5, 2010.
22. Snow DB Jr, Stettin GD; Medco. Healthcare 2020. Drug Trend Report. 2011;13:1–117.
23. Berkrot B. Reuters. Update 1-FDA to review Teva biosimilar of Amgen's Neupogen. February 2, 2010. Available at http://www.reuters.com/article/2010/02/02/teva-neupogen-idUSN0224168320100202. Accessed October 27, 2011.
24. Insmed Inc. Insmed announces first human bioequivalence data for a follow-on biologic by a U.S. company. July 10, 2008. Available at http://investor.insmed.com/releasedetail.cfm?ReleaseID=320665. Accessed October 26, 2011.
25. Weise M, Bielsky M-C, De Smet K, et al. Biosimilars-why terminology matters. Nature Biotechnol. 2011;29(8):690–693.


Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).
Management of antipsychotic medication polypharmacy
June 13th 2013Within our healthcare-driven society, the increase in the identification and diagnosis of mental illnesses has led to a proportional increase in the prescribing of psychotropic medications. The prevalence of mental illnesses and subsequent treatment approaches may employ monotherapy as first-line treatment, but in many cases the use of combination of therapy can occur, leading to polypharmacy.1 Polypharmacy can be defined in several ways but it generally recognized as the use of multiple medications by one patient and the most common definition is the concurrent use of five more medications. The presence of polyharmacy has the potential to contribute to non-compliance, drug-drug interactions, medication errors, adverse events, or poor quality of life.
Medical innovation improves outcomes
June 12th 2013I have been diagnosed with stage 4 cancer of the pancreas, a disease that’s long been considered not just incurable, but almost impossible to treat-a recalcitrant disease that some practitioners feel has given oncology a bad name. I was told my life would be measured in weeks.

