
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Emerging risk factors and risk markers for cardiovascular disease: Looking beyond NCEP ATP III
This review discusses emerging risk factors for CVD, including hs-CRP, lipoprotein(a), homocysteine, fibrinogen, homocysteine, and coronary artery calcification.



Key Points
Abstract
Cardiovascular disease (CVD) continues to be the leading cause of death in the United States. The current standard of care is to treat patients to a target low-density lipoprotein (LDL) cholesterol level, as recommended by the National Cholesterol Education Program (NCEP) Adult Treatment Panel (ATP) III. Unfortunately, despite treating patients to their respective LDL goals, many patients continue to suffer cardiovascular events. Evidence of risk factors for CVD beyond lipids is mounting in the literature. NCEP ATP III now recommends testing some patients for emerging cardiovascular risk factors, such as levels of C-reactive protein (CRP), fibrinogen, coronary artery calcification (CAC), homocysteine, lipoprotein(a), and small, dense LDL. Agents that may be used to ameliorate these emerging risk factors include statins, niacin, fibric acid derivatives, bile acid sequestrants, ezetimibe, thiazolidinediones, and aspirin. Selection of the most appropriate therapy requires an understanding of the function each emerging risk factor plays in atherosclerosis and the mechanisms of therapy. (Formulary. 2009;44:237–247.)
Despite recent advances in the management of dyslipidemia, hypertension, and diabetes mellitus, cardiovascular disease (CVD) remains the leading cause of death in the United States.1 Current preventative strategies for CVD focus on treating patients to specific lipid goals as identified in the National Cholesterol Education Program (NCEP) Adult Treatment Panel (ATP) III and its 2004 update.2,3 NCEP ATP III bases these goals on a patient's risk factor(s) for CVD, such as hypertension (HTN), low high-density lipoprotein (HDL) cholesterol levels, family history of premature CVD, age, and tobacco use. These risk factors are specified because of their well-established association with CVD.

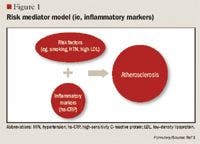
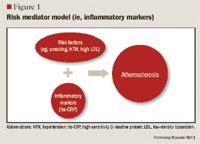
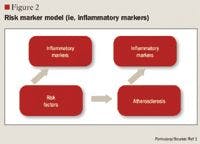
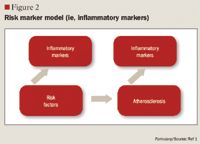



LIPOPROTEIN SUBFRACTION ANALYSIS AND INSULIN RESISTANCE
Lipids (cholesterol esters and triglycerides) are transported through the body in complexes known as lipoproteins. Lipoproteins vary in size and density and are designated as LDL, HDL, and very low-density lipoprotein (VLDL). Laboratory-reported LDL concentration actually represents a measurement of not just one lipoprotein, but rather a spectrum of LDL particles ranging in size and density.6



It has been well established that smaller, more dense LDL particles confer a greater CV risk, and that by altering LDL pattern, this risk may be reduced.6 These data support the idea that LDL quality (ie, size and density) may matter more than LDL quantity.8 The predominance of small, dense LDL (sdLDL) particles has been linked to a 3- to 7-fold increased risk of coronary artery disease (CAD) when compared with the risk associated with a preponderance of large LDL particles.8 A partial explanation may be that sdLDL particles are taken up more readily by the arterial tissue and are also more susceptible to oxidation than larger, more buoyant LDL particles.8,9
The quintessential pattern B patient is the patient with insulin resistance, and this patient often presents with metabolic syndrome. Metabolic syndrome is a constellation of risk factors that increase a patient's risk for CVD and diabetes.2 Factors indicative of metabolic syndrome include abdominal obesity, high triglycerides (TG), low HDL, impaired fasting glucose, and HTN. This TG-rich lipoprofile is associated with an increase in sdLDL particles.8 sdLDL levels may also be significantly elevated in men, in postmenopausal women, and in patients with a genetic predisposition, increased age, or high-carbohydrate diet.8



In 1 study, immediate-release niacin (niacin IR) 3,000 mg/d (n=48) was studied versus atorvastatin 10 mg/d (n=53) in patients with total cholesterol levels >200 mg/dL and TG levels of 200 to 800 mg/dL.7 Both agents reduced sdLDL by a significant amount (44.3% with atorvastatin and 35.1% with niacin IR), but niacin IR increased the number of larger LDL particles by 75%, whereas atorvastatin reduced the number of these particles by 9.8%. Additionally, niacin IR shifted more patients from pattern B to pattern A than did atorvastatin (41% vs 12%, respectively). Another trial compared 120 men with baseline TG levels >190 mg/dL who were treated with either lovastatin 20 mg twice/d plus colestipol 10 g 3 times/d or lovastatin 20 mg twice/d plus niacin IR 4,000 mg/d.12 Patients treated with lovastatin plus niacin IR demonstrated a 20% increase in pattern A dyslipidemia versus a 6% increase in those patients treated with lovastatin plus colestipol.
Fibrates have also been demonstrated to alter LDL particle size. In 1 study, healthy, nonobese patients with type IIA dyslipidemia were randomized to receive fenofibrate 200 mg/d (n=36) or pravastatin 20 to 40 mg/d (n=43) for 16 weeks.13 Fenofibrate-treated patients demonstrated a significant increase in LDL particle size. The proportion of fenofibrate-treated patients with sdLDL was reduced from 69.4% to 30.6% (P<.05), whereas the proportion of pravastatin-treated patients with sdLDL was reduced from 81.4% to 72.1% (not significant). A similar study compared fenofibrate 200 mg/d with atorvastatin 10 mg/d in patients with type 2 diabetes mellitus and mixed dyslipidemia.14 In this study, atorvastatin reduced LDL by 29% and lowered sdLDL but had no significant effect on increasing the number of larger, more buoyant LDL particles. Conversely, fenofibrate reduced LDL by 11% but also reduced sdLDL by 31% (P<.05) and increased intermediate-density LDL by 36%.
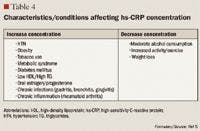
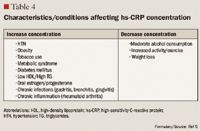
HS-CRP
The role of inflammation in CVD has been well documented over the past decade. Inflammation has been demonstrated to be involved in all stages of atherosclerosis, from initiation and growth to plaque rupture.15 hs-CRP levels are increased in patients with acute inflammatory events such as infection, autoimmune disorders, and the chronic inflammation caused by atherosclerotic plaque formation. The correlation between hs-CRP and CV events is so strong that hs-CRP retains an independent association with coronary events even after adjusting for age, total cholesterol (TC), HDL cholesterol, tobacco use, body mass index (BMI), diabetes, HTN, exercise level, and family history of CVD.5,15 It has been noted that CRP levels are up-regulated in the presence of atherosclerotic plaques, promoting LDL cholesterol uptake by macrophages and facilitating the recruitment of circulating monocytes to plaque sites, thus furthering atherogenesis.16
Despite this strong evidence linking elevated hs-CRP to CVD, it remains unknown whether CRP is a risk mediator or a risk marker.1 One recent study by Elliot et al17 was undertaken to answer this question. This study investigated the association of genetic loci with CRP levels and the risk of CVD. The researchers identified a single-nucleotide polymorphism (SNP) in the CRP gene that was strongly associated with CRP levels, with each minor allele associated with a 21% decrease in CRP concentration. Despite a prediction of a 6% CVD risk reduction based on this decrease in CRP, this SNP was not associated with CVD. From these results, the authors concluded that although CRP is elevated in CVD, it does not appear to be a cause of CVD.
Other studies, however, have demonstrated that hs-CRP levels may have prognostic value for predicting a cardiovascular event in patients with acute coronary syndrome (ACS), those undergoing coronary revascularization procedures using percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG), and, most robustly, in patients with no known history of CVD.1,4,15,18 One of the first studies to link hs-CRP to increased CVD risk was the Physicians' Health Study.16 This study divided patients into quartiles by baseline hs-CRP, with ≤0.55 mg/L as the lowest level and ≥2.11 mg/L as the highest level. The Physicians' Health Study indicated that patients with hs-CRP levels in the highest quartile are at 2 times the risk of stroke, 3 times the risk of MI, and 4 times the risk of peripheral arterial disease compared with those with hs-CRP levels in the lowest quartile. These findings were strengthened by data from the Women's Health Study, which demonstrated that women with LDL <130 mg/dL but with hs-CRP levels in the highest tertile ( >3 mg/L) have 3 times the risk of experiencing coronary events in the future.19 More recently, a cohort of 5,067 patients without CVD underwent testing to determine if CRP was predictive of CV and coronary events after a mean of 12.8 years.20 The authors observed that the strongest association between CV events and a single biomarker was the association between these events and CRP, which demonstrated a multivariable-adjusted HR of 1.19 (95% CI, 1.07-1.32) for each standard deviation increment in CRP. Another recent publication demonstrating the correlation between CVD and hs-CRP is the Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) study.4 The JUPITER study enrolled 17,802 patients with a normal LDL (<130 mg/dL) but an elevated hs-CRP (>2 mg/L) and compared treatment with rosuvastatin 20 mg/d versus placebo. The study was halted after 1.9 years of follow-up. Treatment with rosuvastatin produced a 44% decrease in the composite risk of MI, stroke, arterial revascularization, hospitalization for unstable angina, and death from CV causes, further demonstrating that hs-CRP is closely related to CVD.




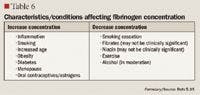
FIBRINOGEN
Fibrinogen has been identified as an emerging risk factor for CVD.2 Like hs-CRP, fibrinogen is an acute phase reactant. The conversion of fibrinogen to fibrin, which is triggered in response to vascular or tissue injury, is the final step in the clotting cascade. Fibrinogen has also been linked to CVD through other mechanisms, such as decreasing blood viscosity; increasing platelet aggregation; causing vasoconstriction at sites of vessel wall injury; and increasing cell adhesion, chemotaxis, and proliferation.5 However, there are no therapies that have been demonstrated to lower fibrinogen concentrations enough to reduce CV risk.5,32,33
Fibrinogen antigen is measured by immunonephelometry, a test normally used to diagnose dysfibrogenemias, which may lead to bleeding disorders. A fibrinogen antigen level >350 mg/dL is considered elevated. Because fibrinogen is an acute phase reactant, levels should not be tested in patients with active bleeding, those with acute infection, or those who have received blood transfusions in the previous 4 weeks.10


EFFECT OF LP(A)
Lp(a) is an LDL-like particle. The protein constituent consists of lipo-protein(a), or Apo(a), linked to apolipoprotein B, the standard lipoprotein of the LDL particle.5 The Lp(a) particle is similar in composition and density to a normal LDL particle, but has an added deleterious effect because of its structural similarity to plasminogen.36 In addition to having thrombotic properties, Lp(a) is also 40% cholesterol by mass; therefore, it also has the ability to cause atherosclerosis.5
Levels of Lp(a) are classified into quartiles, which correlate to increasing risk for CVD. An Lp(a) level <20 mg/dL is considered desirable, whereas a level of 20 to 30 mg/dL is borderline, 31 to 50 mg/dL is high risk, and >50 mg/dL is very high risk. Lp(a) is measured by enzyme-linked immunosorbent assay (ELISA) testing and may vary based on ethnicity, with Asian and Caucasian patients having the highest levels.10
Of the currently known emerging CV risk factors, Lp(a) seems to have the strongest hereditary link, but not all studies have demonstrated an increased risk of CVD associated with increased Lp(a).5,37,38 A meta-analysis of 27 prospective studies that included >5,400 patients with CVD demonstrated that patients with Lp(a) measurements in the top third of the population are at a 70% increased risk of death from CVD versus those with measurements in the bottom third.39 In contrast, the Genetic Epidemiology Network of Arteriopathy (GENOA) study included 765 men and women and demonstrated no correlation between Lp(a) and coronary calcification, with correlation coefficients of 0.055 (P=.33) and 0.052 (P=0.27) in men and women, respectively.38 Beyond the fact that data seem to be conflicting regarding the importance of Lp(a) as a risk factor for CVD, there are also currently no good treatment options for elevated Lp(a) levels.


HOMOCYSTEINE
One of the most hotly debated risk factors for CVD continues to be homocysteine. Homocysteine is formed as a by-product of the metabolism of the essential amino acid methionine and is then remetabolized by multiple enzymes that use folic acid, cobalamin (vitamin B12), and pyridoxine (vitamin B6) as substrates.5 Although homocysteine has no known biologic function, it is a highly reactive amino acid that can increase CVD risk if levels are elevated. One meta-analysis calculated an odds ratio of 1.7 (95% CI, 1.5–1.9) for CAD in patients with elevated homocysteine levels.44 Several mechanisms have been described to explain the correlation between elevated homocysteine and CVD. Homocysteine is thought to injure endothelial cells, alter platelet activity, inhibit vasodilation, and cause thrombogenesis.45,46 Increased homocysteine levels likely increase CVD risk when augmented by other well-established risk factors (eg, smoking, HTN, elevated LDL) but may not increase risk when not associated with these other risk factors.5 One of the most common causes of hyperhomocysteinemia is a gene mutation in the 5,10-methylenetetrahydrofolate reductase (MTHFR) enzyme, which is responsible for metabolizing homocysteine. This genetic abnormality can lead to a 25% increase in homocysteine levels, especially in the setting of low folate intake.5
Plasma homocysteine is measured by competitive immunoassay and should be tested when patients are in the fasting state, as higher levels are noted after eating. Levels >10 mcmol/L are thought to place patients at higher risk of CVD. Very high homocysteine levels (>100 mcmol/L) are typically caused by homozygous cystathionine-beta-synthase (CS) deficiency, which occurs in 1 per 300,000 live births. Patients with the MTHFR variant will likely have moderate elevations in homocysteine levels (16–30 mcmol/L).10



Conversely, another trial assigned 5,522 patients with documented vascular disease or diabetes to treatment with folic acid 2.5 mg, vitamin B6 50 mg, and vitamin B12 1 mg or placebo.48 The occurrence of death from CV causes, MI, or stroke was 18.8% in the treatment group and 19.8% in the placebo group (P=.41). Patients in both groups had a mean baseline homocysteine level of 12.2 mcmol/L; the treatment group demonstrated a decrease from baseline of 2.4 mcmol/L in plasma homocysteine level.
CORONARY ARTERY CALCIFICATION
Whereas the aforementioned tests may have some predictive value for CVD, the presence of coronary calcium indicates that CVD is present and can be used to predict the risk of a CV event. Coronary artery calcification (CAC) is virtually absent in normal arteries. CAC occurs early in atherosclerotic plaque formation in the second and third decades of life and progresses with further growth of the plaque. CAC seems to be most useful in identifying higher-risk asymptomatic individuals with an intermediate CVD risk based on family history and Framingham risk score.49,50 CAC is predictive of death or MI at 3 to 5 years and is independently predictive when controlling for standard risk factors.49
CAC is measured using an electron-beam computed tomography (EBCT) scan, which allows for the acquisition of 1.5- to 3-mm sections. Each identified lesion is assigned a score of 1 to 4 based on the size of the lesion; the largest plaques receive a score of 4. The total coronary calcium score is then determined by adding up each lesion score for all sequential slices. CAC scores <100 are associated with a low probability (0.4%) of annual death or MI, whereas scores of 100 to 399 indicate that a moderate amount of plaque is present (annual risk of death or MI, 1.3%).50 A CAC score ≥400 puts a patient at a similar risk to that associated with diabetes or PAD, 2 well-known coronary heart disease risk equivalents.2 Because CAC is a marker of plaque formation, there is no treatment directed to reduce CAC. Rather, the occurrence of a high CAC score would result in a recommendation to be more aggressive in other methods to decrease plaque (eg, lipid-lowering therapy), as this high score indicates that CVD is already present.50
RECOMMENDED SCREENING
With the availability of new tests and accumulating literature support regarding the importance of these emerging risk factors, providers and healthcare systems may be tempted to consider screening everyone for these risk factors. Additionally, as the results of these trials reach the lay press, educated patients may begin to request testing for emerging risk factors. Universal screening is not cost-effective, however, and it does not add clinical benefit in all situations.






CONCLUSION
CVD remains the leading cause of mortality in the United States despite advances in lipid management. Although a reduction in LDL cholesterol remains the major therapeutic target, it is important to remember that high LDL is not the only cause of CVD. Searching for nontraditional risk factors may be clinically useful in a certain subset of patients. The risk:benefit ratio of emerging risk factor testing and therapy must be evaluated on a case-by-case basis, and the importance of patient education and medication adherence must be stressed. Look for prescribing patterns to change as more data are published on these important risk factors.
Dr Hoffmann and Dr Tucker are clinical pharmacists, Blanchard Valley Medical Associates, Findlay, Ohio. Dr Parker is an assistant professor of pharmacy practice, College of Pharmacy, University of Findlay, Ohio.
Acknowledgment: The authors would like to thank Dr David Meier and Dr Catherine Waggoner-Meier for their assistance in preparing this manuscript.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. Field KM. Effect of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on high-sensitivity C-reactive protein levels. Pharmacotherapy. 2005;25:1365–1377.
2. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285:2486–2497.
3. Grundy SM, Cleeman JI, Merz CNB, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines [erratum in Circulation. 2004;110:763]. Circulation. 2004;110:227–239.
4. Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207.
5. Hackam DG, Anand SS. Emerging risk factors for atherosclerotic vascular disease: A critical review of the evidence. JAMA. 2003;290:932–940.
6. Rizzo M, Berneis K. Low-density lipoprotein size and cardiovascular risk assessment [erratum in QJM. 2007;100:147]. QJM. 2006;99:1–14.
7. McKenney JM, McCormick LS, Schaefer EJ, Black DM, Watkins ML. Effect of niacin and atorvastatin on lipoprotein subclasses in patients with atherogenic dyslipidemia. Am J Cardiol. 2001;88:270–274.
8. Rizzo M, Berneis K. Who needs to care about small, dense low-density lipoproteins? Int J Clin Pract. 2007;1949–1956.
9. Rajman I, Maxwell S, Cramb R, Kendall M. Particle size: The key to the atherogenic lipoprotein? QJM. 1994;87:709–720.
10. LabCorp Provider. Available at http://https://www.labcorp.com/. Accessed July 9, 2009.
11. Backes JM, Gibson CA. Effect of lipid-lowering drug therapy on small-dense low-density lipoprotein. Ann Pharmacother. 2005;39:523–526.
12. Brown BG, Zambon A, Poulin D, et al. Use of niacin, statins, and resins in patients with combined hyperlipidemia. Am J Cardiol. 1998;81:52B–59B.
13. Lemieux I, Laperriere L, Dzavik V, Tremblay G, Bourgeois J, Despres J. A 16-week fenofibrate treatment increases LDL particle size in type IIA dyslipidemic patients. Atherosclerosis. 2002;162:363–371.
14. Frost RJ, Otto C, Geiss HC, Schwandt P, Parhofer KG. Effects of atorvastatin versus fenofibrate on lipoprotein profiles, low-density lipoprotein subfraction distribution, and hemorheologic parameters in type 2 diabetes mellitus with mixed hyperlipoproteinemia. Am J Cardiol. 2001;87:44–48.
15. Pearson TA, Mensah JA, Alexander RW, et al. Markers of inflammation and cardiovascular disease. Circulation. 2003;107:499-511.
16. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men [erratum in N Engl J Med. 1997;337:356]. N Engl J Med. 1997;336:973–939.
17. Elliott P, Chambers JC, Zhang W, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48.
18. Torzewski M, Rist C, Mortensen RF, et al. C-reactive protein in the arterial intima: Role of C-reactive protein receptor-dependent monocyte recruitment in atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20:2094–2099.
19. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843.
20. Melander O, Newton-Cheh C, Almgren P, et al. Novel and conventional biomarkers for prediction of incident cardiovascular events in the community. JAMA. 2009;302:49–57.
21. Muhlestein JB, May HT, Jensen JR, et al. The reduction of inflammatory biomarkers by statin, fibrate, and combination therapy among diabetic patients with mixed dyslipidemia: The DIACOR (Diabetes and Combined Liquid Therapy Regimen) study. J Am Coll Cardiol. 2006;48:396–401.
22. McKenney J. Niacin for dyslipidemia: Considerations in product selection. Am J Health Syst Pharm. 2003;60:995–1005.
23. Prasad K. C-reactive protein (CRP)-lowering agents. Cardiovasc Drug Rev. 2006;24:33–50.
24. Devaraj S, Autret B, Jialal I. Effects of colesevelam hydrochloride (WelChol) on biomarkers of inflammation in patients with mild hypercholesterolemia. Am J Cardiol. 2006;98:641–643.
25. Ballantyne CM, Houri J, Notarbartolo A, et al; Ezetimibe Study Group. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: A prospective, randomized, double-blind trial. Circulation. 2003;107:2409–2415.
26. Sager PT, Capece R, Lipka L, et al. Effects of ezetimibe coadministered with simvastatin on C-reactive protein in a large cohort of hypercholesterolemic patients. Atherosclerosis. 2005;179:361–367.
27. Dandona P. Effects of antidiabetic and antihyperlipidemic agents on C-reactive protein. Mayo Clin Proc. 2008;83:333–342.
28. Mohanty P, Aljada A, Ghanim H, et al. Evidence for a potent anti-inflammatory effect of rosiglitazone. J Clin Endocrinol Metab. 2004;89:2728–2735.
29. Brunzell JD, Marcovina S, Yu D, et al. Rosiglitazone reduces novel biomarkers of cardiovascular disease in subjects with type 2 diabetes mellitus already on statin therapy [abstract 1124-192]. J Am Coll Cardiol. 2004;43(suppl 2):504A.
30. Ikonomidis I, Andreotti F, Economou E, Stafanidis C, Toutouzas P, Nihoyannopoulos P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation. 1999;100:793–798.
31. Feldman M, Jialal I, Devaraj S, Cryer B. Effects of low-dose aspirin on serum C-reactive protein and thromboxane B2 concentrations: A placebo-controlled study using a highly sensitive C-reactive protein assay. J Am Coll Cardiol. 2001;37:2036–2041.
32. Pazzucconi E, Mannucci L, Mussoni L, Gianfranceschi G, et al. Bezafibrate lowers plasma lipids, fibrinogen and platelet aggregability in hypertriglyceridemia. Eur J Pharmacol. 1992;43:219–223.
33. Guyton JR, Blazing MA, Hagar J, et al. Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Arch Intern Med. 2000;160:1177–1184.
34. Green D, Foiles N, Chan C, Schreiner PJ, Liu K. Elevated fibrinogen levels and subsequent subclinical atherosclerosis: The CARDIA study. Atherosclerosis. 2009;202:623–631.
35. Meade TW. Fibrinogen and cardiovascular disease. J Clin Pathol. 1997;50:13–15.
36. Parker DL. Cardiovascular risk: What should we measure? US Pharm. 2009;34:25–27.
37. Moliterno D, Jokinen EV, Miserez AR, et al. No association between plasma lipoprotein(a) concentrations and the presence or absence of coronary atherosclerosis in African-Americans. Arterioscler Throm Vasc Biol. 1995;15:850–855.
38. Kullo IJ, Bailey KR, Bielak LF, et al. Lack of association between lipoprotein(a) and coronary artery calcification in the Genetic Epidemiology Network of Arteriopathy (GENOA) study. Mayo Clin Proc. 2004;79:1258–1263.
39. Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease. Meta-analysis of prospective studies. Circulation. 2000;102:1082–1085.
40. Pan J, Van JT, Chan E, Kesala RL, Lin M, Charles MA. Extended-release niacin treatment of the atherogenic lipid profile and lipoprotein(a) in diabetes. Metabolism. 2002;51:1120–1127.
41. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanik ML, Jackson RD, et al; Writing Group for the Women's Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333.
42. Duriez P, Dallongeville J, Fruchart JC. Lipoprotein(a) as a marker for coronary heart disease. Br J Clin Pract. 1996;77A:54–61.
43. Anderson JL. Lipoprotein-associated phospholipase A2: An independent predictor of coronary artery disease events in primary and secondary prevention. Am J Cardiol. 2008;101:23F–33F.
44. Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. JAMA. 1995;274:1049–1057.
45. Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet and cardiovascular diseases: A statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999;99:178–182.
46. Temple ME, Luzier AB, Kazierad DJ. Homocysteine as a risk factor for atherosclerosis. Ann Pharmacother. 2000;34:57–65.
47. Hackam DG, Peterson JC, Spence JD. What level of plasma homocyst(e)ine should be treated? Effects of vitamin therapy on progression of carotid atherosclerosis in patients with homocyst(e)ine levels above and below 14 micromol/L. Am J Hypertens. 2000;13:105–110.
48. The Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease [erratum in N Engl J Med. 2006;355:746]. N Engl J Med. 2006;354:1567–1577.
49. Greenland P, Bonow RO, Brundage BH, Budoff MJ, Eisenberg MJ, Grundy SM, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: A report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000 Expert Consensus Document on Electron Beam Computed Tomography) developed in collaboration with the Society of Atherosclerosis Imaging and Prevention and the Society of Cardiovascular Computed Tomography. J Am Coll Cardiol. 2007;49:378–402.
50. Pletcher MJ, Tice JA, Pignone M, McCulloch C, Callister TQ, Browner WS. What does my patient's coronary artery calcium score mean? Combining information from the coronary artery calcium score with information from conventional risk factors to estimate coronary heart disease risk. BMC Med. 2004;2:31.
51. Change:Healthcare. Available at: http://https://www.changehealthcare.com/. Accessed July 9, 2009.

FDA Approves Combination Therapy for Pulmonary Arterial Hypertension
March 25th 2024J&J’s Opsynvi is single-tablet combination of macitentan, an endothelin receptor antagonist, and tadalafil, a PDE5 inhibitor. It will be priced on parity with Opsumit, which is also a J&J product to treat patients with PAH.




FDA Issues Complete Response Letter for Onpattro in Heart Failure Indication
October 9th 2023Alnylam Pharmaceuticals will no longer pursue this indication of Onpattro and will instead on focus on a label expansion for Amvuttra, which is in phase 3 development to treat patients with cardiomyopathy of ATTR amyloidosis.


