
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
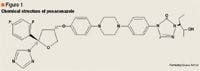
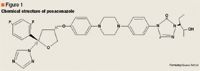
Focus on posaconazole: A novel triazole antifungal for the treatment of invasive fungal infections
Serious infections due to the Aspergillus and Candida species and other filamentous fungi have emerged as an increasing cause of infectious morbidity and mortality in the United States and globally. The most notable explanation for this increase is a rise in the number of immunocompromised patients due to advances in transplantation, the emergence and prevalence of Acquired Immunodeficiency Syndrome (AIDS), and an increase in the number of invasive surgical procedures. 1-3 Treatment of these infections with currently available standard antifungal agents such as amphotericin B, itraconazole, and fluconazole still results in an unacceptably high associated mortality. 3. Furthermore, many of these antifungal agents have limitations, including dose-limiting toxicity, drug-drug interactions, and fungal resistance. 4-10



The problem of resistance has been exacerbated by a limited antifungal armamentarium. For example, resistance to flucytosine among various yeasts is the major cause of treatment failure with this agent.4 Azole resistance among Candida species has been well documented; moreover, fluconazole, a commonly used azole antifungal, has no activity against molds.5-7 In addition, this class of agents may have clinically significant interactions with drugs metabolized by the cytochrome P450 enzyme system (CYP450), creating particular concern for immunocompromised patients since this population commonly receives many of these drugs and simultaneously is at greatest risk for fungal infections.8 Voriconazole, the most recent azole to be approved, has addressed some resistance issues but is still affected by increases in fluconazole minimum inhibitory concentrations (MIC) due to efflux pumps encoded by the genes, FLU1 and MDR1.6,9 Additionally, voriconazole is not without adverse events.10 Although amphotericin B resistance to date remains rare, concerns about serious adverse events with the deoxycholate formulation, most notably nephrotoxicity and infusion-related reactions, have limited the usefulness of this drug.11,12 Lipid formulations of amphotericin B have demonstrated similar efficacy with fewer adverse events, but are very costly and thus often saved for those patients unable to tolerate the conventional formulation. Finally, caspofungin, a novel echinocandin antifungal, has in vitro activity against both Candida and Aspergillus species with little resistance, but clinical experience with this agent is still emerging, and it is not available in an oral formulation.
Therefore, there is a critical need for newer antifungals that are more potent, effective, and safe than the currently available agents. Posaconazole (Schering-Plough) is a new second-generation, broad-spectrum triazole under FDA review for the treatment of invasive fungal infections refractory to other antifungals. Approval in the United States is anticipated in early 2005. A thorough evaluation of its pharmacology, antifungal spectrum of activity, clinical trial results, safety, and tolerability is discussed in this review.


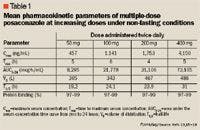
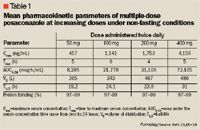
Posaconazole is orally bioavailable, with maximum concentrations reached approximately 6 to 10 hours post-dose.13,15 The absorption of posaconazole is linear through the clinically useful dose of 400 mg every 12 hours; absorption can be saturated at higher doses. Like itraconazole, posaconazole absorption is greatly affected by food.16,17 There is an approximate 2-fold increase in absorption of the oral suspension when taken with food, and a 4-fold increase in absorption with a high-fat meal. Multiple-dose studies demonstrated that splitting the dose increased the total amount of drug absorbed.15 A tablet formulation that is unaffected by food was tested during phase 1 studies; however, it was found to have a 35% lower bioavailability compared with the oral suspension.16
Posaconazole is slowly metabolized to several minor inactive metabolites, the most significant of which is the glucuronide conjugate, termed M8, accounting for 17.5% to 27.6% of the administered dose.13 This limited metabolism is mediated predominantly through phase 2 biotransformations via UPD-glucuronosyltransferase (UGT) enzyme pathways, and less through the oxidative pathways of the CYP450 system.20 Importantly, unlike other azole antifungals, posaconazole administration results in parent drug plasma concentrations that exceed those of its metabolites over a 24-hour period.13 Thus, the active drug predominates, enabling prolonged microbiological exposure.

Employers Face Barriers With Adopting Biosimilars
March 1st 2022Despite the promise of savings billions of dollars in the United States, adoption of biosimilars has been slow. A roundtable discussion among employers highlighted some of the barriers, including formulary design and drug pricing and rebates.






