
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Generic inhaled corticosteroids: Current status and what to expect next
Tracking and preparing for the approval of generic products is 1 of the key components of proactive formulary management. Unfortunately, forecasting FDA approval of the first generic inhaled corticosteroid products is difficult.
Maria M. Ousterhout, PharmD
Dr Ousterhout is clinical consultant pharmacist, University of Massachusetts Medical School Clinical Pharmacy Services, Shrewsbury, Mass.
The author reports no financial disclosures as related to products discussed in this article.
Abstract:
Tracking and preparing for the approval of generic products is 1 of the key components of proactive formulary management. Unfortunately, forecasting FDA approval of the first generic inhaled corticosteroid products is difficult. This is because this class of agents faces significant challenges in both the development and approval processes. Several scientific challenges have been identified in demonstrating bioequivalence of a generic inhaled corticosteroid to the reference branded product. FDA has recognized these challenges and is encouraging the development of collaborative solutions among the various stakeholders. Currently, an estimation of when these generic products are expected to enter the market is not available; however, conversations between various academic, industry, and regulatory stakeholders are ongoing to help determine a clear path forward for these products. This article provides an overview of the current state of generic inhaled corticosteroids, outlines the challenges that generics manufacturers face, and discusses the implications for potential market entry.
ESTIMATING GENERIC ENTRY
In a managed care environment, it is important to stay ahead of changes in the pharmaceutical market and proactively develop appropriate formulary management strategies. Besides keeping current with drugs that are new to the marketplace, it is important to know when brand-name pharmaceutical products with a large portion of the market share are expected to become available generically. This awareness can help managed care decision-makers develop optimal drug management strategies that ensure patients have continuous access to the most appropriate and least costly therapeutic options. Unfortunately, determining when the switch from brand to generic will occur can be difficult to forecast. One class of medications that has been particularly problematic to track along this path is inhaled corticosteroids (ICs). This class of medications has faced barriers in the development and approval pathways that have hindered the approval of a generic IC.
THE GENERIC DRUG APPROVAL PATHWAY
To be reviewed by FDA, generic pharmaceutical manufacturers must submit an abbreviated new drug application (ANDA) for the generic for which approval is sought.1,2 The application used for generic approval is considered abbreviated because it does not require duplication of data obtained through clinical trials that established safety and efficacy of the reference product. These applications must instead contain information to support the claim that the generic and reference products are equivalent.1 A generic product must be comparable to the reference branded product in dosage form, strength, route of administration, product stability, quality, performance characteristics, and intended use.1,3,4 Manufacture of the generic product is held to the same standards of FDA’s good manufacturing practice regulations that are applied to the making of the reference product.4
In place of the clinical tests required for approval of a novel pharmaceutical agent, generics makers must demonstrate that the product is pharmaceutically equivalent and bioequivalent to the reference product. This is required in order to prove that the generic product will have similar action to the reference product.1,3 For products to be considered pharmaceutically equivalent, both products need to contain the same active ingredient, be of the same strength, and utilize the same dosage form.3 For a generic product to be considered bioequivalent, it must be delivered at the same rate and extent to the target site of action when it is administered at the same dose and under the same conditions as the reference.5 In other words, the product must deliver the same amount of active ingredient into the bloodstream over the same amount of time when compared with the reference product.1
The Code of Federal Regulations (21 CFR 320.24) lists 5 possible approaches for demonstrating bioequivalence.6 The first is to conduct in vivo studies that compare drug or active metabolite concentrations in the plasma or urine. The second is to use in vivo study to compare the pharmacologic effect of the drug. The third is to conduct a clinical trial to evaluate and document the safety and efficacy of the drug. Fourth is to utilize in vitro methods that are considered acceptable to FDA. Lastly, the regulation states any other method deemed appropriate by FDA can be used to demonstrate bioequivalence.
For many products, demonstrating bioequivalence is straightforward and uncomplicated because pharmacokinetic measures can be used. The most common way to demonstrate the safety and efficacy of a generic compared with the reference product is to compare plasma drug concentrations.3 To do so requires measuring the time that it takes the generic product to reach the bloodstream and to enter systemic circulation in a small group of healthy volunteers.1 This information can be used to determine the rate of absorption (the bioavailability) of the generic product, which can then be compared with the reference product.1 Unfortunately, not all drug products lend themselves to such a straightforward comparison. When commonly used methods for demonstrating bioequivalence cannot be applied, there is a significant challenge to the development and subsequent approval of a generic product.3 One class of medications for which this is the case are ICs.
FDA REQUIREMENTS FOR APPROVAL OF GENERIC ICs
FDA has specific requirements that must be met for a generic IC product to be considered bioequivalent to a reference product. According to communication with the Division of Drug Information at FDA’s Center for Drug Evaluation and Research (CDER), generic manufacturers must conduct both a local delivery pharmacodynamic dose-response study to establish that delivery to the lungs is equivalent and a pharmacokinetic study to demonstrate that systemic exposure with the generic product is equivalent to that of the reference product. Manufacturers must also show that the generic product has a comparable metered dose inhaler (MDI) or dry powder inhaler (DPI) device design. Lastly, the excipients used in the generic product must be both qualitatively and quantitatively the same as those used in the reference product.
CHALLENGES OF DEVELOPING GENERIC ICs
The suggested methods for demonstrating bioequivalence have been problematic for companies seeking to develop a generic IC, for many reasons. First, it has not been shown that drug concentrations in the plasma or serum reflect the amount of drug that is delivered to the lung, the target site of action.7 Determining a dose-response relationship for ICs has proven especially difficult because these agents exhibit a flat dose-response. This means that it is possible for a brand and proposed generic equivalent to deliver different amounts of active drug without it being easily detectable. Without a clear method for evaluating the impact of delivering different doses, it is challenging to develop generic products that can deliver levels of the active ingredient that are comparable to the reference product.8
Use of clinical trials is another option for demonstrating bioequivalence. Although comparative clinical trials may have established bioequivalence with orally inhaled products in the past, these trials are not ideal for the assessment of generic ICs. This is due in part to great variability between patients, to the amount of time and resources required to complete trials, and because of the difficulty in interpreting the findings of these studies.8
Besides demonstrating bioequivalence of the drug, developing a generic IC must take into account the design of the drug-delivery system. There are 2 main types of delivery systems used for ICs: MDI and DPI. An MDI utilizes a forceful burst of drug to deliver the dose. This is dependent on the drug formulation, the canister the product is stored in, and the release valve and corresponding actuator device. In contrast, DPIs tend to be diverse in design and instructions for use. This type of device relies on the patient’s inhalation to aerosolize the drug product. Differences in inspiratory flow among patients can result in differences in the particle-size distribution in the aerosol. Unfortunately, there is a lack of accepted in vitro methodologies to assess the differences in individual device designs. In addition, the lack of a correlation between the in vitro performance of a device and in vivo performance makes it difficult to properly assess the impact of variations in device design on bioequivalence of a potential generic.8
FDA acknowledges that there are several challenges posed by the requirements set forth for generic ICs and has conceded that it is difficult to appropriately demonstrate bioequivalence on these measures. Moreover, because of the lack of a formal FDA-issued guidance describing the best approach to designing trials necessary to meet these requirements, FDA has not received many applications for these products regardless of the patent status on the reference product.3 Based on communication with FDA’s CDER, until there is published FDA guidance, the Office of Generic Drugs (OGD) has advised that generic manufacturers can conduct their own studies to determine the methodology most appropriate to demonstrate bioequivalence, specifically the dose-response relationship. It is also recommended, however, that manufacturers discuss planned pharmacodynamic studies with OGD before initiating them, to ensure the design is adequate.
FDA CRITICAL PATH INITIATIVE
FDA’s Critical Path Initiative focuses on the challenges involved in the development of new innovator drugs, devices, and biologics. In 2007, FDA released a document titled “Critical Path Opportunities for Generic Drugs” that outlined specific areas of interest within the realm of generic drug development and approval. FDA posted the document on its website to bring these challenges to the attention of key stakeholders and interested parties. The goal of FDA’s report and listing of these challenges is to promote discussion of the issues among stakeholders and potentially lead to the coming together of various groups to develop collaborative solutions. Ultimately, the report was produced to help facilitate the development and approval processes for generic products.3
One of the 4 areas of opportunity identified by FDA is the development of methods for assessment of bioequivalence of locally acting drugs, such as topical and inhalation products. As previously stated, assessing and demonstrating bioequivalence for locally acting inhaled products is impeded by several scientific challenges. Determining bioequivalence is problematic because the drug concentration in the plasma or that is demonstrated via in vitro dissolution testing may not be an appropriate surrogate of the pharmacologic activity of these agents.3
After carefully evaluating the hurdles generic ICs are expected to overcome, FDA identified several specific challenges that require a closer look so that all interested parties can develop a solution and find the best path forward for these products. The first of these is developing molecular-level imaging techniques that can quantify the amount of drug that is at the site of action. These techniques can be used to help validate information from in vitro tests or to help identify biomarkers that could be used for future testing of ICs. Second, given the complex nature of the delivery systems used with ICs, there is a need to identify key device performance and formulation variables to help FDA establish products that are within the proper equivalency limits and quality specifications. Third, consideration should be given to evaluating products that utilize different formulations. Previously, FDA had requested that all nasal and inhaled products have formulations that are both qualitatively and quantitatively identical to the reference product. FDA states that it is important that the impact of qualitative and quantitative differences be explored to determine if there are other acceptable paths forward with respect to this requirement.3
Additionally, there is a need to develop pharmacodynamic study designs that could use forced expiratory volume in 1 second (FEV1) as a clinically significant surrogate end point in clinical trials utilizing a cross-over design. Cross-over design would help reduce the number of test subjects that are required for evaluations of inhaled products, since typically studies require a very large patient population to help overcome the high levels of within- and between-subject variability that is inherent in the disease states these agents are used to treat.3 Unfortunately, this type of study design is not as strong as a randomized controlled trial and comes with some limitations. In these trials, it is possible that the order in which interventions are studied can affect patient outcomes. The effects of the first intervention may not completely wear off by the time the second intervention is administered. So while cross-over studies may be useful in reducing the size of the population required, it is important to consider the limitations inherent in this design.
Another aspect of clinical trial design to consider is the development of a study design for patients with chronic obstructive pulmonary disease (COPD). This is important because COPD consists of chronic bronchitis, emphysema, and chronic asthma with patients potentially presenting with symptoms that overlap between these classifications. All 3 manifestations of COPD have different degrees of symptom reversibility in response to treatment. Lastly, since many inhalation products are available as single entities as well as combination products, special consideration should be given to the development of a generic combination product. One of the key steps to demonstrating bioequivalence is the ability to prove that local delivery, of both components, is equivalent. Unfortunately, because of the pathophysiology of the disease states these agents treat, both ingredients have an initial impact on the patient’s FEV1 but the impact may vary as time passes, depending on the specific ingredients in the formulation. A nitric oxide end point, another surrogate marker that may be used, is only affected by the corticosteroid component of inhaled products, so possibly combining this with an initial FEV1 should be considered to allow for the determination of bioequivalence of both active ingredients.3 This biomarker has been shown to be decreased in a dose-dependent manner after administering clinically relevant doses of IC. Unfortunately, the use of this marker comes with some problems as well. For example, patients without asthma or COPD may still have elevated levels of nitric oxide and it is speculated that the response to an IC may be related to the baseline level of nitric oxide, prior to drug administration. Currently, the use of this biomarker is being evaluated in an exploratory study sponsored by FDA’s Critical Path Initiative.8
COLLABORATION TO OVERCOME THESE CHALLENGES
The Product Quality Research Institute (PQRI) is 1 of the groups actively involved in working toward a solution to the problems that generics manufacturers face in developing generic ICs. PQRI is a nonprofit consortium with representation from research organizations, the pharmaceutical industry, and FDA’s CDER. The purpose of this group is to collaborate to develop and disseminate information that can help advance both drug development and quality.9 Based on a proposal from the American Association of Pharmaceutical Scientists, specifically the Inhalation and Nasal Technology Focus Group, PQRI sponsored a workshop on “Demonstrating Bioequivalence of Locally Acting Orally Inhaled Drug Products.” The March 2009 workshop included participants and experts from generics manufacturers, pharmaceutical companies, various regulatory bodies, and from academia in the United States, Europe, and other parts of the world.8
The purpose of the workshop was to review, discuss, and consider possible recommendations for demonstrating bioequivalence of locally acting, inhaled products. The workshop also focused on discussing and considering regulatory guidance for new drug applications, ANDAs, and any necessary post-approval changes. The presentations and discussions from the 2-day workshop addressed a wide range of topics, including in vitro approaches to demonstrating bioequivalence, biomarker strategies, imaging techniques, in vivo approaches to establishing local delivery equivalence, and device design similarity. Representatives from FDA summarized the problems facing manufacturers of generic ICs and acknowledged that FDA is stretched too thin to develop solutions to these problems on their own. FDA welcomed participation from the various areas of drug development represented by the workshop attendees.8
At the conclusion of the workshop a list of unanswered questions and points requiring clarification was developed. Specifically, the attendees determined that additional clarification is required regarding the ideal study designs for assessing generic ICs, specifically how to select a patient population for products that can be utilized for both asthma and COPD. The attendees also asserted that clarification is needed regarding the definition of sameness as it relates to device design for ICs. More information is required to determine the therapeutic impact of changes in device design. Questions arose regarding what would be considered acceptable differences in the product instructions for use or corresponding labeling, and whether small innovations to device design (such as reducing the number of actuations required to prime an MDI) affect the ability to establish sameness. The outcome of the presentations and discussions suggested that currently there is not a clear pathway for demonstrating bioequivalence of generic ICs.8
In April 2010, PQRI sponsored another workshop entitled “Role of Pharmacokinetics in Establishing Bioequivalence for Orally Inhaled Drug Products.” A summary of the presentations and discussions held during the workshop was published in 2011.10 As with the earlier workshop, representation from the pharmaceutical industry, various regulatory bodies throughout the world, as well as the realm of academia was included. Discussions revolved around using pharmacokinetics data as the basis for determining in vivo equivalence and suggested a possible approach that would pair this information with in vitro testing. While the 2009 workshop highlighted the areas that would require further research and outlined what may be a long path ahead, the summary of proceedings of the April 2010 workshop demonstrate the ongoing efforts across the various groups of stakeholders to continue the work toward finding a solution.10
WHAT’S NEXT?
Table 1 lists the dates that generic IC products could be expected to enter the market, based on patent expiration information alone. This information may serve as a benchmark for estimating when generic products will be permissible, but it is important to keep in mind that these dates do not account for delays that could result from the challenges described above.11 Unfortunately, the path toward a generic approval in this category of medications is more complicated than it is for other products and it is currently unclear if these patent expiration dates will be useful in estimating market entry for these agents.

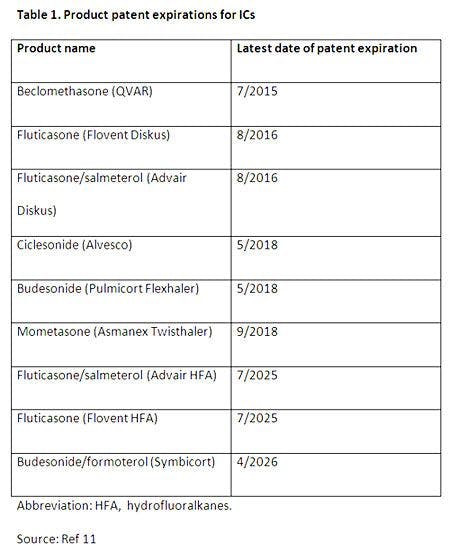

Adding to the uncertainty about when generics might appear on the shelves, it has been reported that Teva, one of the world’s largest makers of generic drugs, recently decided to shift its focus from developing a generic version of fluticasone and salmetero (Advair) to developing a branded competitor that could potentially compete with the product instead.12 Teva believes it will be able to submit this product for FDA review in 2014 and explained that it is pursuing this path because they believe overcoming regulatory hurdles to gain FDA approval of a generic IC are so great that it is not possible to develop an interchangeable product. Sandoz, the generic unit of Novartis, has also decided to terminate development of a generic Advair.12 Furthermore, Medco released a document that lists anticipated generics over the next several years and has recently begun leaving out all IC-containing products with a note that FDA has not yet finalized the standard for demonstrating bioequivalence of these products. The report also states that currently there is no estimated date for generic approval of these agents.13
As of June 2011, e-mail communication from the Division of Drug Information at FDA’s CDER indicated that FDA has not yet released any updated guidance or new information related to generic ICs. This communication included a referral to the PQRI workshop on demonstrating bioequivalence of locally acting, orally inhaled drug products for additional information. A detailed summary of PQRI workshop presentations and discussions can be found in the Journal of Aerosol Medicine and Pulmonary Drug Delivery.8
CONCLUSION
Manufacturers of generic ICs face significant challenges in the development and approval processes. These challenges and unanswered questions underscore the need for continued collaborative efforts between FDA and various interested groups from the pharmaceutical industry and academic and research worlds. It is important that representatives from each of these areas work together to develop solutions that will be optimal for all involved. Such collaborations will be critical in determining the path forward for these agents.
It is also important for healthcare decision-makers, including those in managed care, to keep current with any developments in this process. In addition to monitoring discussions regarding the potential paths toward approval of generic ICs, it is also critical to continue to track patent expirations associated with these products. This should include keeping a close watch on not only individual drug products and chemical entities, but also any patents on delivery devices. Pending finalization of requirements for demonstrating equivalence between delivery devices, the patents on the delivery devices for existing branded ICs may play an important role in dictating when a generic equivalent can enter the market. There currently is no specific estimate for when generic IC products will become available, but managed care decision-makers can utilize this time to evaluate current management strategies for the IC class and to prepare for any future changes that generic ICs may bring.
REFERENCES
- FDA. Abbreviated new drug application (ANDA): generics. Silver Spring, MD: FDA, US Department of Health and Human Services; 2011.
- Drug Price Competition and Patent Term Restoration Act of 1984, S. 1538, 98th Cong. (1984).
- FDA Office of Generic Drugs, Office of Pharmaceutical Science, Center for Drug Evaluation and Research. Critical path opportunities for generic drugs. Silver Spring, MD: FDA, US Department of Health and Human Services; 2007.
- FDA. Generic drugs: questions and answers. Silver Spring, MD: FDA, US Department of Health and Human Services; 2010.
- Bioavailability and bioequivalence requirements: definitions, 21 C.F.R. Sect. 320.21 (2002). Available at http://www.law.cornell.edu/cfr/text/21/320. Accessed October 10, 2011.
- Bioavailability and bioequivalence requirements: Types of evidence to establish bioavailability or equivalence, 21 C.F.R. Sect. 320.24 (1992). Available at: http://edocket.access.gpo.gov/cfr_2001/aprqtr/pdf/21cfr320.24.pdf.
- Singh GJP. Evolution of regulatory and scientific paradigms for establishing equivalence of systemic exposure from orally inhaled drugs: current status and possible challenges. Respiratory Drug Delivery. 2010;1:249–260.
- Adams WP, Ahrens RC, Chen ML, et al. Demonstrating bioequivalence of locally acting orally inhaled drug products (OIPs): workshop summary report. J Aerosol Med Pulm Drug Deliv. 2010;23:1–29.
- Product Quality Research Institute. PQRI Website. http://www.pqri.org/. Updated June, 2011. Accessed June 15, 2011.
- O’Connor D, Adams WP, Chen ML, et al. Role of pharmacokinetics in establishing bioequivalence for orally inhaled drug products: workshop summary report. J Aerosol Med Pulm Drug Deliv. 2011;24:119–135.
- FDA, Center for Drug Evaluation and Research. Orange Book, 31st ed. Approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Updated April, 2011. Accessed June 14, 2011.
- Hirschler B. Glaxo breathes easier as threat to Advair recedes. Reuters Website. http://www.reuters.com/article/2010/11/05/us-glaxo-advair-idUSTRE6A42ZH20101105. Updated November 5, 2010. Accessed June 17, 2011.
- Medco. Anticipated first time generics. Medco About Generics Website. http://www.medcohealth.com/medco/corporate/home.jsp?articleID=CorpAboutGenerics. Updated January, 2011. Accessed June 17, 2011.


Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).
Management of antipsychotic medication polypharmacy
June 13th 2013Within our healthcare-driven society, the increase in the identification and diagnosis of mental illnesses has led to a proportional increase in the prescribing of psychotropic medications. The prevalence of mental illnesses and subsequent treatment approaches may employ monotherapy as first-line treatment, but in many cases the use of combination of therapy can occur, leading to polypharmacy.1 Polypharmacy can be defined in several ways but it generally recognized as the use of multiple medications by one patient and the most common definition is the concurrent use of five more medications. The presence of polyharmacy has the potential to contribute to non-compliance, drug-drug interactions, medication errors, adverse events, or poor quality of life.
Medical innovation improves outcomes
June 12th 2013I have been diagnosed with stage 4 cancer of the pancreas, a disease that’s long been considered not just incurable, but almost impossible to treat-a recalcitrant disease that some practitioners feel has given oncology a bad name. I was told my life would be measured in weeks.

