
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Gout: Overview and newer therapeutic developments
The main objectives of gout therapy are to treat the acute attack, provide prophylaxis to prevent flares, and prevent complications associated with the deposition of urate crystals in tissues. Obesity, alcohol intake, and certain foods and medications can contribute to hyperuricemia and should be identified. Pharmacologic management remains the mainstay of treatment for chronic gout and acute attacks.



Key Points
Abstract
Gout is a chronic, progressive disease that is increasing in prevalence. It results from the deposition of urate crystals caused by the overproduction or underexcretion of uric acid. The disease is often, but not always, associated with elevated serum uric acid levels. The phases of gout include asymptomatic hyperuricemia, acute gouty arthritis, intercritical gout, and chronic advanced gout. Diagnosis is based on the identification of uric acid crystals in joints, tissues, or body fluids. The main objectives of therapy are to treat the acute attack, provide prophylaxis to prevent flares, and prevent complications associated with the deposition of urate crystals in tissues. Obesity, alcohol intake, and certain foods and medications can contribute to hyperuricemia and should be identified. Pharmacologic management remains the mainstay of treatment for chronic gout and acute attacks. (Formulary. 2010;45:84-90.)
Gout is the most common inflammatory arthritis disorder in the United States; currently there are an estimated 3 to 5 million sufferers and its prevalence appears to be increasing.1 Gout affects about 2% of men (>30 years) and women (>50 years) and accounts for about 3.9 million annual physician visits, with more than two-thirds occurring at the primary care practitioner's office.2,3

PATHOGENESIS
Uric acid, or urate, is the end product of purine degradation in humans.4 As the genetic exon for the enzyme uricase cannot be expressed in humans, uric acid cannot be enzymatically converted into allantoin (a more water-soluble product). Without uricase, uric acid accumulates as the final product of catabolism of the purines. The amount of urate in the body depends on the balance between dietary intake, synthesis, and excretion.
Hyperuricemia is defined as serum uric acid at a concentration >6.8 mg/dL, the saturation point at which urate crystallizes in biological fluids at normal body temperature (98.6°F).5 Hyperuricemia is believed to result from underexcretion (~90%) or overproduction (~10%) of uric acid.6 Although hyperuricemia necessarily precedes gout, some patients with hyperuricemia never experience a single attack of gouty arthritis. In general, the higher a patient's serum uric acid level, the greater the likelihood that the patient will develop gout.7 Because it is difficult to predict which patients with asymptomatic hyperuricemia will develop articular gout, asymptomatic hyperuricemia is typically not treated.
Gout develops when hyperuricemia leads to the deposition of monosodium urate (MSU) crystals in joints and soft tissues. After MSU crystals are deposited around joints, local factors such as trauma or irritation, previous disease, and lower temperatures may initiate the release of crystals into the joint space, triggering the inflammation of an acute gouty attack. These attacks can be self limiting (3–14 days), but crystals often remain in the joint and cause low-grade inflammation between acute attacks.7
RISK FACTORS
The risk factors for the development of gout have been frequently identified, explained, and reviewed in the literature. Nonmodifiable risk factors include being a male or a postmenopausal female, genetic influence, end-stage renal disease, and resulting from major organ transplantations. Gout's prevalence increases with age, from 1.8/1,000 in those aged less than 45 to 30.8/1,000 in those aged more than 65.8 Hyperuricemia and gout have been linked to other disease states including hypertension, metabolic syndrome, cardiac disease, renal disease, and truncal obesity. Certain dietary factors and medications have been linked to gouty attacks and should be minimized in patients with a history of gout. Foods that have been implicated in causing gout are red organ meats, seafood (especially shellfish), foods containing high-fructose corn syrup, and high alcohol intake (especially beer).8 Medications, particularly thiazide diuretics and cyclosporine administered to transplant patients, have been implicated in gouty attacks.8
CLINICAL PRESENTATION
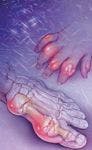
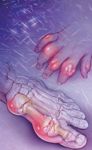
The classical presentation of gout is podagra, that is, inflammation of the first metatarsophalangeal (MTP) joint of the foot. This joint is eventually affected in approximately 90% of patients with gout. Joints in the lower extremities are the most common site of an initial acute gout attack, but gout may also affect bursae and tendons.7 Gouty attacks begin abruptly (usually at night) and reach maximum intensity within 8 to 12 hours. The joints are red, hot, and exquisitely tender; even a bed sheet on the swollen joint can be uncomfortable.
Four clinical manifestations are seen in gout: asymptomatic hyperuricemia, acute gouty arthritis, intercritical gout, and chronic advanced gout.7 At serum concentrations higher than 6.8 mg/dL, urate crystals may start to deposit. During this first phase of gout, asymptomatic hyperuricemia, urate deposits may directly contribute to organ damage. After sufficient urate deposits have developed and some traumatic event triggers the release of crystals into the joint space, a patient will suffer an acute gout attack and move into the second phase, acute gouty arthritis. This involves acute inflammation of the joint caused by urate crystallization, which is described as the "flare." The interval between gouty attacks is the third phase, intercritical gout. When the crystal deposits continue to accumulate, patients develop stiff and swollen joints leading to the final phase, chronic advanced gout. This includes long-term complications of uncontrolled hyperuricemia such as chronic arthritis and tophi. Tophi are nodular masses of uric acid crystals deposited in different soft-tissue areas of the body. This fourth phase of gout is uncommon because it can be avoided with proper medication treatment.7
Historically, a disease of affluent, middle-aged, or older men with overindulgent lifestyles, gout has now become more generalized, affecting more women and a wider range of socioeconomic groups. A number of factors have been proposed to explain the increasing prevalence of gout in the United States. Increased longevity may be a key factor because prevalence is a function of both disease incidence and disease duration. Other contributing factors may be the increased prevalence of hypertension and metabolic syndrome, increased use of diuretics and low-dose aspirin, dietary trends, change in demographics, increased prevalence of end-stage renal disease, and increases in organ transplantation.9
DIAGNOSIS
The American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) have established evidence-based guidelines for diagnosing gout.10-12 Identifying MSU crystals in synovial fluid remains the gold standard for diagnosing gout, but these guidelines can be helpful for making a preliminary diagnosis without crystal confirmation. A number of laboratory and other tests may assist with the diagnosis and/or differential diagnosis of gout. Serum urate levels are important, but 50% of patients suffering an acute attack will have normal serum urate levels.13 Baseline laboratory tests should also include a complete blood count, urinalysis, blood urea nitrogen, and serum creatinine. Renal function is important because many gout treatments are contraindicated, require dose alterations in renally impaired patients, or require more stringent follow-up.
PHARMACOTHERAPY



TREATMENT OF ACUTE GOUT

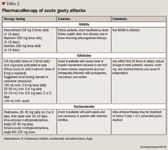
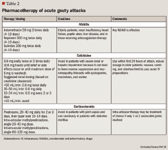
NSAIDs are the preferred therapy for the treatment of patients without complications. Indomethacin is commonly used for gout, but others including ibuprofen, naproxen, sulindac, piroxicam, and ketoprofen are also effective for the treatment of acute gout.15 Maximum doses should be given after onset of symptoms or at time of diagnosis and continued for 24 hours after complete resolution of the acute attack, then tapered off over 2 to 3 days. All NSAIDs can have serious gastrointestinal (GI) side effects including bleeding and ulceration, which can limit their use. These drugs should be used with caution in patients with a history of peptic ulcer disease, congestive heart failure, and chronic renal disease.
Colchicine, an antimitotic drug derived from the roots of the herb colchicum autumnale, is one of the oldest treatments of gout. It is most beneficial when it is given in the first 12 to 36 hours of an attack. It exerts its effects by inhibiting the phagocytosis of uric acid and blocking the release of chemotactic factors. Colchicine has anti-inflammatory activity but no analgesic activity. Although colchicine is effective in treating acute gout, 80% of patients experience GI side effects, including nausea, vomiting, and diarrhea at standard therapeutic doses.16
EULAR recommends colchicine at a dosage of 0.6 mg 3 times daily, but there is currently no consensus on the most appropriate dosing regimen.12 A multicenter, randomized, double-blind, placebo-controlled trial compared the efficacy and safety of high-dose colchicine regimen (1.2-mg loading dose followed by 0.6 mg/h for 6 hours to total of 4.8 mg) to a low-dose colchicine regimen (1.2-mg loading dose followed by 0.6 mg/h for 1 hour to total of 1.8 mg followed by 5 placebo doses hourly) in 184 patients.17 Low-dose colchicine was shown to be as effective as high-dose colchicine, but was associated with a lower incidence of GI adverse effects. It was this lower dosage regimen that formed the basis for the recent approval (July 2009) of single-ingredient oral colchicine by FDA.18
Intravenous (IV) colchicine is available but has a narrow therapeutic-toxicity level. Improper intravenous colchicine therapy has been associated with bone marrow suppression, myoneuropathy, renal failure, disseminated intravascular coagulation, tissue necrosis from extravascular extravasation, and death.19
Corticosteroids are also used to treat acute attacks and can be administered orally, or by intra-articular or intramuscular injection. They are a useful alternative when patients have renal and/or GI contraindications to other treatments, but should be avoided in patients with joint sepsis and used with caution in diabetic patients.14
Adrenocorticotropic hormone (ACTH) has also been used in the treatment of acute gout. A dose of 40 IU given intramuscularly and repeated every 8 to 24 hours, as needed, is as effective as indomethacin at a dose of 50 mg, 3 times daily.20 Although parenteral ACTH is effective, it may require several repeated injections and cannot be used in patients who have recent prior use of systemic steroids, since the action of ACTH requires an unsuppressed adrenal axis.
Very acute gout with other renal manifestations of extreme hyperuricemia can arise during the unusual tumor lysis syndrome.21 Tumor lysis syndrome is characteristically seen during induction therapy of acute leukemia, often in children.
A recombinant, non-mammalian urate oxidase (uricase), known as rasburicase, was approved by FDA in 2002 to treat hyperuricemia associated with the tumor lysis syndrome.22 An IV infusion of recombinant rasburicase will enzymatically oxidize uric acid to 5-hydroxyisourate, which is then converted to allantoin in the body, as noted previously.4 Rasburicase infusions are approved to be administered daily for up to 5 days as a treatment or prophylaxis of the tumor lysis syndrome. Singular infusions of rasburicase have been utilized off-label to address uncontrolled uric acid levels with or without accompanying gout.23
TREATMENT OF INTERCRITICAL GOUT
As previously discussed, after the phase of acute gouty arthritis the patient returns to an asymptomatic phase of the disease, intercritical gout. During this phase, medications should be assessed and dietary education regarding purine-rich foods (which contribute to higher serum uric acid levels) should be discussed. Patients also should be educated about weight loss and healthy lifestyle habits.
At this point, the decision to initiate prophylactic hyperuricemic therapy is discussed. Generally, patients with hyperuricemia and recurrent attacks, chronic gout, tophi, gouty arthritis, or nephrolithiasis should be treated. There is debate on when to initiate urate-lowering therapy, but it has been found that urate-lowering therapy is cost effective for patients who have 2 or more attacks per year.24
Low-dose colchicine or low-dose NSAIDs should be used for prophylaxis only with concurrent urate-lowering agents, as chronic prophylaxis with either of these agents does engender their own specific adverse effects. These medications are used for prophylaxis until the serum urate concentration is stable at a desired level and the patient has been free from acute gouty attacks for 3 to 6 months. Acute gouty flare-up may occur when prophylaxis is discontinued, but most patients are able to remain on urate-lowering agents alone.14,15
TREATMENT OF CHRONIC GOUT
After the complete resolution of an acute gout attack and the initiation of prophylactic therapy, options are available to reduce the likelihood of recurrence. The frequency of subsequent acute attacks of gout increases with time. Approximately 60% of patients will have a second attack within the first year, and 78% have a second attack within 2 years. Only 7% do not have a recurrence within a 10-year period.25 The therapeutic goal of urate-lowering therapy is to promote dissolution of the urate crystals and to prevent crystal formation. The main treatment goal is the reduction of serum uric acid to 6.0 mg/dL or lower. Two main drug classes for lowering urate are uricosuric agents and xanthine oxidase inhibitors. It is recommended that patients who overproduce urate should be treated with xanthine oxidase inhibitors and patients who underexcrete it despite nearly normal creatinine clearance levels should be treated with uricosurics.14 Medications for urate-lowering therapy must be taken for the rest of a patient's life.
Uricosuric agents decrease the serum uric acid level by increasing renal excretion. Probenecid is the only uricosuric agent available in the United States. It works at the level of the proximal tubule by blocking reabsorption of filtered uric acid. Probenecid should be initiated at a dosage of 250 mg twice daily and increased as needed up to 3 g/d to achieve a serum urate level of ≤6.0 mg/dL.
Uricosuric agents should be avoided in patients with a history of nephrolithiasis and are ineffective when given to patients with renal insufficiency. The major side effects include rash, GI intolerance, and uric acid stone formation. One limitation of uricosuric medications is that low doses of aspirin can block their uricosuric effects.26
In contrast to uricosuric agents, xanthine oxidase inhibitors are effective in patients who overproduce or underproduce uric acid.19 Allopurinol had been the mainstay of urate-lowering therapy for about 40 years until febuxostat was approved in 2009.27
Allopurinol is a structural analogue of hypoxanthine that competitively inhibits the conversion of hypoxanthine to xanthine and the conversion of xanthine to uric acid. It therefore decreases uric acid levels in the serum and urine and increases hypoxanthine and xanthine levels.19 Allopurinol is often started at 100 mg/d, with the daily dosage increased in 100-mg increments every 2 to 4 weeks. The usual dosage range for allopurinol is 200 to 300 mg/d for mild gout and 400 to 600 mg/d for cases of moderate-to-severe gout.8 The allopurinol dose must be adjusted in patients with renal impairment.

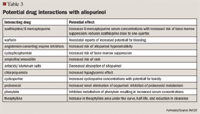
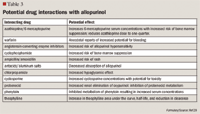
Febuxostat is a nonpurine, selective xanthine oxidase inhibitor, which was approved February 2009 for the chronic management of hyperuricemia in patients with gout.27 The recommended initial dose is 40 mg once daily. If the target serum urate concentration of <6 mg/dL is not reached after 2 weeks, dosage may be increased to 80 mg once daily.
Because febuxostat is chiefly metabolized or conjugated by the liver, no dosage adjustment is needed for patients with mild-to-moderate renal or hepatic impairment.27 Adverse effects for febuxostat include elevations of liver enzymes, rash, diarrhea, and headache. Febuxostat is contraindicated in patients treated with azathioprine, mercaptopurine, or theophylline, according to the manufacturer.30 However, the manufacturer has not conducted clinical drug interaction studies with any of these drugs. The warning regarding azathioprine, mercaptopurine, and theophylline is based on the fact that each of these drugs is at least partially metabolized by xanthine oxidase.
The manufacturer has reported that febuxostat has a higher rate of cardiovascular events (ie, cardiovascular deaths, nonfatal myocardial infarction, and nonfatal strokes) as compared with allopurinol.30 Long-term extension studies looked at these cardiovascular events with either 80-mg febuxostat or oral allopurinol. The long-term cardiovascular incidences were 0.97 event with febuxostat and 0.58 event with allopurinol, each event indexed per 100 patient-years of therapy. Importantly, each of these cardiovascular events had widely overlapping statistical confidence intervals.30
In a phase 3, multicenter, double-blind, 6-month, randomized controlled trial of 2,269 gout patients, allopurinol (300 mg or 200 mg daily for patients with chronic kidney disease) was compared with febuxostat (40 mg or 80 mg daily) for efficacy in reducing serum uric acid levels to <6.0 mg/dL.30,31 Febuxostat at a dose of 40 mg had efficacy comparable to allopurinol, with significantly greater efficacy at a dose of 80 mg (P<.001). In patients with chronic kidney disease, febuxostat 40 mg and 80 mg demonstrated significantly greater efficacy than did allopurinol 300 mg and 200 mg.
In a 5-year extension study of this trial, 95 (83%) of 114 patients had a serum uric acid level <6 mg/dL, and the mean serum uric acid levels were reduced 45% to 59% from baseline.32 By the end of the extension study, the number of patients experiencing gout flares had declined to zero.
As compared with allopurinol, febuxostat can be used without a dose reduction in patients with mild-to-moderate renal impairment, does not interrupt the metabolism of many other concomitant drugs, and does not cause the hypersensitivity reactions that are dose limiting with allopurinol. However, febuxostat has been associated with increased cardiovascular events when compared with allopurinol, but not to a statistically significant degree.30
Febuxostat is the first novel medication approved for gout in more than 40 years, but it is comparatively more expensive. Nevertheless, febuxostat is a welcomed alternative therapy for gout, particularly when there are complicating issues such as allergy or inefficacy with allopurinol.
CONCLUSION
Gout is a disease with increasing prevalence. Hyperuricemia can lead to urate supersaturation, urate crystal formation, and ultimately gout, which if left untreated can lead to permanent joint damage. Acute attacks commonly present as severe pain in a single joint, most often the first MTP joint. The clinical goal in managing chronic or refractory gout is reducing the severity and frequency of attacks. NSAIDs or other anti-inflammatory drugs may reduce the time of resolution of the initial attack, but long-term use of urate-lowering medications is needed to minimize the chance of recurrence.
Dr Hilaire is a faculty member of the University of Wyoming School of Pharmacy in Laramie, and the Fort Collins Family Medicine Residency Program in Colorado. She is a graduate of Duquesne University School of Pharmacy in Pittsburgh, PA. Dr Wozniak is a faculty physician at the Fort Collins Family Medicine Residency Program in Fort Collins, CO, and she is a graduate of Loyola University Medical School in Chicago.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25.
2. Choi HK, Curhan G. Gout: epidemiology and lifestyle choices. Curr Opin Rheumatol. 2005;17:341–345.
3. Krishnan E, Lienesch D, Kwoh CK. Gout in ambulatory care settings in the United States. J Rheumatol. 2008;35:498–501.
4. Hahn PC, Edwards NL. Management of hyperuricemia. In: Koopman WJ, Moreland LW, eds. Arthritis and Allied Conditions. 15th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:2341–2355.
5. Kippen I, Klinenberg JR, Weinberger A, Wilcox WR. Factors affecting urate solubility in vitro. Ann Rheum Dis. 1974;33:313–317.
6. Pittman JR, Bross MH. Diagnosis and management of gout. Am Fam Physician. 1999;59:1799–1806.
7. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75(suppl 5):S5–S8.
8. Mandel DA, Simkin PA. Gout: update on pathogenesis, diagnosis, and treatment. New Develop Rheum Dis. 2007:20–25.
9. Bieber JD, Terkeltaub RA. Gout: on the brink of novel therapeutic options for an ancient disease. Arthritis Rheum. 2004;50:2400–2414.
10. Wallace SL, Robinson H, Masi AT, Decker JL, McCarty DJ, Yü TF. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895–900.
11. Zhang W, Doherty M, Pascual E, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1301–1311.
12. Zhang W, Doherty M, Pascual E, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1312–1324.
13. Weaver AL, Cheh MA, Kennison RH. How PCP education can impact gout management: the gout essentials. J Clin Rheumatol. 2008;14(suppl 5);S42–S46.
14. Dore RK. Gout: what primary care physicians want to know. J Clin Rheumatol. 2008;14(suppl 5):S47–S54.
15. Wallace SL, Singer JZ. Therapy in gout. Rheum Dis Clin North Am. 1988;14:441–457.
16. Wallace SL, Singer JZ. Review: systemic toxicity associated with the intravenous administration of colchicine – guidelines for use. J Rheumatol. 1988;15:495–499.
17. Terkeltaub R, Furst D, Bennett K, Kook K, Davis M, Bethesda S. Low dose (1.8 mg) vs high dose (4.8 mg) oral colchicine regimens in patients with acute gout flare in a large, multicenter, randomized, double-blind, placebo-controlled, parallel group study. [abstract] Arthritis Rheum. 2008;58(suppl):S897–S880.
18. Colchicine (marketed as Colcrys). Available at: http://www.fda.gov./Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm174596.htm. Accessed February 4, 2010.
19. Schlesinger N. Management of acute and chronic gouty arthritis: present state-of-the-art. Drugs. 2004;64:2399–2416.
20. Axelrod D, Preston S. Comparison of parenteral adrenocorticotropic hormone with oral indomethacin in the treatment of acute gout. Arthritis Rheum. 1988;31:803–805.
21. Jasek AM, Day HJ. Acute spontaneous tumor lysis syndrome. Am J Hematol. 1994;47:129–131.
22. Elitek (rasburicase) [package insert]. Bridgewater, NJ: Sanofi – Aventis U.S.; 2002.
23. Richette P, Bardin T. Successful treatment with rasburicase of a tophaceous gout in a patient allergic to allopurinol. Nat Clin Pract Rheumatol. 2006:2:338–342.
24. Emmerson BT. The management of gout. N Engl J Med. 1996;334:445–451.
25. Gutman AB. The past four decades of progress in the knowledge of gout, with an assessment of the present status. Arthritis Rheum. 1973;16:431–445.
26. Wortmann RL. Gout and hyperuricemia. Curr Opin Rheumatol. 2002;14:281–286.
27. Drugs@FDA. Rockville, MD: Food and Drug Administration, Center for Drug Evaluation and Research. Febuxostat. Available at: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/. Accessed February 4, 2010.
28. Bohan KH, Russell TM. Gout and hyperuricemia. In: Koda-Kimble MA, Young LY, Kradjan WA, et al, eds. Applied Therapeutics: The Clinical Use of Drugs. 9th ed. Philadelphia, PA: Lippincott Williams and Wilkins;2008:42.1–42.14.
29. Tatro DS, ed. 2008 Drug Interaction Facts. St. Louis, MO: Facts and Comparisons; 2008.
30. Uloric (febuxostat) [package insert]. Deerfield, IL: Takeda Pharmaceuticals North America; 2009.
31. Becker M, Schumacher HR Jr, Espinoza L, et al. A phase 3 randomized, controlled, multicenter, double-blind trial (RCT) comparing efficacy and safety of daily febuxostat (FEB) and allopurinol (ALLO) in subjects with gout. [abstract]. Arthritis Rheum. 2008;58(suppl 9):L11.
32. Schumacher HR Jr, Becker MA, Lloyd E, MacDonald PA, Lademacher C. Febuxostat in the treatment of gout: 5-yr findings of the FOCUS efficacy and safety study. Rheumatology. 2009;48:188–194.





Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).

