
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Hemophilia: Etiology, complications, and current options in management
Hemophilia is a rare congenital bleeding disorder, resulting from a deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B). Deficiency of either of these factors interrupts normal hemostasis resulting in an inability to form a stable fibrin clot to halt bleeding. This article reviews the etiology of hemophilia, available pharmacologic approaches to bleeding episodes, and treatment options in the presence of complications.



Key Points
Abstract
Hemophilia is a rare congenital bleeding disorder, resulting from a deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B). Deficiency of either of these factors interrupts normal hemostasis resulting in an inability to form a stable fibrin clot to halt bleeding. Prompt treatment of bleeding episodes with hemophilia is essential to prevent long-term complications such as joint or muscle damage. The mainstay of treatment is replacement of the deficient factor. In some cases, desmopressin or antifibrinolytics may also be effective. This article reviews the etiology of hemophilia, available pharmacologic approaches to bleeding episodes, and treatment options in the presence of complications. (Formulary. 2010;45:218-227.)


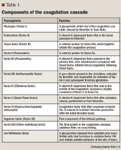
Older explanations of hemostasis described 2 separate pathways for initiation of coagulation-intrinsic and extrinsic-which merged to a common pathway. However, contemporary research suggests that hemostasis is primarily initiated with the release of tissue factor (part of the extrinsic pathway) following vascular injury. Tissue factor then complexes with factor VII, with subsequent conversion of factor VII to activated factor VII (factor VIIa).4 This complex acts locally at the site of injury to activate other factors such as factors X and IX, resulting in the generation of a thrombin "burst." This small amount of thrombin activates platelets and other factors in the intrinsic pathway, including factors VIII and IX. Coagulation then becomes dependent on the actions of factor VIII and IX for additional thrombin generation.6 With sufficient thrombin and other coagulation factors, fibrinogen is converted to fibrin, eventually forming a stable fibrin clot at the site of injury.
HEMOPHILIA A AND B
Disorders of hemostasis can manifest either as excessive bleeding or excessive blood coagulation. Excessive bleeding can occur when proteins or other components critical to coagulation are functionally absent due to either qualitative or quantitative deficiencies.7 Hemophilia A and B develop when there are deficiencies of coagulation factors VIII and IX, respectively.3 Since both factor VIII and IX are needed for thrombin generation and formation of a stable fibrin clot, deficiency of either results in excessive bleeding.
Based on the 2008 Global Survey of the World Federation of Hemophilia, nearly 149,000 individuals worldwide have hemophilia and hemophilia A is the more common factor deficiency.8 In the United States, the number of individuals with hemophilia is estimated at approximately 16,000, with an annual incidence of 1 in 5,000 male births for hemophilia A and 1 in 25,000 male births for hemophilia B.2,9
PATTERNS OF INHERITANCE
Hemophilia A and B are both X-linked recessive disorders resulting from mutations on the factor VIII and factor IX genes located on the X chromosome.10,11 The difference in prevalence between hemophilia A and B is due to the difference in the size of the genes-the factor VIII gene is larger, increasing the chance of a mutation.2 Point mutations are the most commonly found factor VIII and IX gene mutations but other chromosomal aberrations, including deletions, insertions, and rearrangements or inversions have also been identified.10 Male and female offspring of female carriers of a hemophilia gene mutation have a 50% chance of inheriting the mutation; female offspring of males with hemophilia are obligatory carriers because they will always inherit the mutated gene.12 Although females who carry the gene mutation do not usually exhibit spontaneous bleeding, they are likely to have lower levels of clotting factor and may be prone to excessive bleeding following trauma.3,10
DIAGNOSIS



Hemophilia can be severe, moderate, or mild, depending on the degree of factor deficiency. Patients with severe factor deficiency (<1% of normal) experience spontaneous bleeding without apparent trauma. Those with moderate deficiency (1% to 5% of normal) usually do not bleed spontaneously, but may bleed excessively after minor trauma, surgery, or other invasive procedures. Patients with mild deficiency (5% to 40% of normal) experience abnormal bleeding only after significant trauma.13,14
Age at diagnosis is related to the severity of factor deficiency. The median age for diagnosis of severe hemophilia is about 1 month.15 Those with moderate hemophilia are usually diagnosed within the first few years of life but mild hemophilia may be diagnosed much later.3 More than 95% of individuals with hemophilia will be diagnosed by the time they are 15 years of age with approximately 50% having a severe factor deficiency.15
CLINICAL PRESENTATION
Bleeding patterns seen in individuals with hemophilia vary with age.15 Initial bleeding episodes among neonates with severe hemophilia include prolonged bleeding from circumcision and head or intracranial bleeding.3,15 Infants between 1 and 6 months of age may experience soft tissue bleeding or bruising. As mobility increases with age, joint bleeding may be seen. In older children and adults, the joints are the most common site of spontaneous bleeding-referred to as hemarthrosis-and account for 70% to 80% of bleeding episodes. The knees (45%), elbows (30%), and ankles (15%) are the most frequently affected.13 Bleeding can also occur in muscles, soft tissues, or in the central nervous system (CNS).
LONG-TERM COMPLICATIONS
A common long-term complication of hemophilia is permanent damage to the joints (hemarthropathy) caused by repeated bleeding episodes.16,17 Once in the joint, blood can trigger an inflammatory response within the synovial tissue, resulting in tissue damage. Articular cartilage and subchondral bone also are negatively affected by exposure to blood. Damage to the joint tissues can be seen after even short periods of exposure to small amounts of blood. Repeated bleeding into the muscle can also have long-term effects, with muscle and nerve damage potentially leading to contractures.13
TREATMENT
Treatment of hemophilia is comprehensive and focused on preserving both physical health and quality of life of individuals with the disorder.13 The primary goal of treatment is the prevention or cessation of bleeding episodes. Prompt treatment of acute bleeding episodes is essential to minimize long-term complications. Both nonpharmacologic and pharmacologic strategies are used in the treatment of hemophilia.
Nonpharmacologic therapy consists of supportive care. Rest, ice, compression, and elevation (RICE) are important measures for treatment of joint or muscle bleeding episodes.13 Splints, crutches, or casts can be used to allow the affected joint or muscle to rest after a bleeding episode. Cold or ice compresses can help reduce associated inflammation and should be applied for 20 minutes every 4 to 6 hours. Once pain and swelling begin to resolve, physiotherapy can be initiated to maintain joint and muscle function.
Several pharmacologic agents can be used to prevent or treat bleeding episodes. The selection of the most appropriate agent depends on the severity and location of the bleeding, as well as the degree of factor deficiency.
DESMOPRESSIN
Desmopressin acetate (d1-deamino-8-D-arginine vasopressin) is a synthetic analogue of the naturally occurring hormone arginine vasopressin and can be used to treat bleeding in patients with mild hemophilia A.18,19 Although the exact mechanism of action in the treatment of hemophilia A is unknown, administration of desmopressin results in increased plasma levels of both factor VIII and von Willebrand factor, most likely from storage sites in the endothelium. Desmopressin increases factor VIII by 3 to 5 times baseline levels, and the effect is rapid with peak levels occurring 30 to 60 minutes following intravenous administration and 90 to 120 minutes after subcutaneous or intranasal use.18 However, the effects of desmopressin are transient and repeated doses result in tachyphylaxis.18,19 Therefore, the benefits of desmopressin are limited in patients requiring maintenance of adequate factor levels for a prolonged period of time.

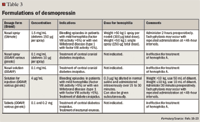
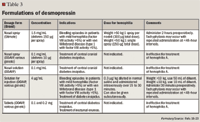
Desmopressin is generally well tolerated, with headache and flushing the most commonly reported adverse events.24 However, there is a risk of water intoxication and hyponatremia, especially in the very young and elderly, if fluid intake is excessive.19,24
ANTIFIBRINOLYTICS
Two antifibrinolytics have been used for treatment of hemophilia-tranexamic acid and aminocaproic acid. These agents help to stabilize a formed fibrin clot by blocking the effects of plasminogen activators.25–27 Antifibrinolytics do not prevent bleeding episodes, but can be effective as adjunctive therapy for mucosal bleedings including epistaxis or those occurring following dental procedures.13 Antifibrinolytics should be avoided in patients with renal bleeding, due to the increased risk of obstructive nephropathy. The usual oral dose of tranexamic acid is 3 or 4 g daily, in divided doses, for 5 to 10 days, depending on the cause of bleeding. For children, tranexamic acid has been given orally at 15 to 25 mg/kg up to 3 times daily. Tranexamic acid 5% solution has also been used topically for bleeding associated with invasive dental procedures.28 Aminocaproic acid is usually given to adults orally as a 5-g dose, followed by 1 g every 8 hours until bleeding subsides.13 The oral dose for children is 50 to 100 mg/kg (maximum 5 g) every 6 to 8 hours. Tranexamic acid is more potent than aminocaproic acid in terms of receptor binding to block the effects of plasminogen activators and is more commonly used.
FACTOR CONCENTRATES
For both hemophilia A and B, replacement of the deficient factor with a factor concentrate is the treatment of choice for most bleeding episodes.3 Some patients with mild hemophilia A may respond to desmopressin unless bleeding is severe or life-threatening, but for those with moderate to severe hemophilia, factor replacement is necessary.
The goal of therapy with factor concentrates is to raise the circulating factor levels high enough to achieve hemostasis. The needed factor level, dose, and duration of therapy are dependent on the site and severity of bleeding.3 Desired factor levels are given as a percentage of normal. Guidelines from the World Federation of Hemophilia recommend an initial desired factor VIII level of 40% to 60% for joint and most muscle bleeding, and 80% to 100% for iliopsoas muscle, throat or neck, CNS, or gastrointestinal bleeding.13 The duration of therapy can range from 1 to 2 days for joint bleeding and up to 21 days for life-threatening bleeding. Similar desired factor levels and duration of therapy are used for patients with hemophilia B.
The dose of factor VIII concentrates is based on in vivo recovery-the rise in plasma factor VIII levels for each unit of factor administered.13,29 On average, plasma factor VIII levels are increased by about 2% (2 U/dL) for each unit of factor per kilogram given intravenously.
The initial dose of factor VIII can be calculated by:
weight (kg) x (percent desired factor level–percent baseline factor level) x 0.5
For example, a 50-kg patient with bleeding into the knee joint would need factor levels raised to 60% of normal. Since most patients with spontaneous bleeding have little endogenous factor (baseline factor = 0%), the calculated dose would be:
50 kg x 60 (% desired level) x 0.5 = 1,500 units of factor VIII
This can be followed with a maintenance dose, which is based on the half-life of factor VIII, amounting to 50% of the initial dose given every 12 hours.30 Factor VIII may also be given as a continuous infusion. This method of administration has several advantages over bolus administration, including a reduction in factor requirements and avoidance of excess peak levels or trough levels below that needed for hemostasis.31 Comparisons between intermittent bolus injections of factor VIII and a continuous infusion have reported a decrease in clearance, lower total factor use, and fewer bleeding complications with the continuous infusion.32,33
For factor IX, each unit per kilogram raises plasma factor IX levels by about 1% (1 U/dL). For the same patient, the dose for factor IX can be calculated as:
50 kg x 60 (% desired level) = 3,000 units of factor IX
However, with recombinant factor IX, in vivo recovery is lower (0.7 to 0.8 U/dL for each unit of factor/kg), and higher doses are needed compared to plasma-derived factor IX products.30 A maintenance dose, usually 50% of the initial dose, can then be given every 24 hours because factor IX has a longer half-life than factor VIII.
For both factors, subsequent dosing can be determined by in vivo recovery for that patient and the patient's clinical status; in some cases, a second loading dose may be needed.29 Both factor VIII and factor IX can be given by slow intravenous push (<3 mL/min for adults and <100 U/min for children).13
ADVERSE EVENTS
Factor concentrates are generally well tolerated, with headache reported in about 2% of patients.30,34–46 Hypersensitivity reactions manifesting with hives, urticaria, wheezing or chest tightness, hypotension, and anaphylaxis; and thrombotic complications, primarily with high doses, have also been reported. However, the 2 main concerns with factor replacement are transmission of pathogenic viruses or prions and the development of inhibitors.
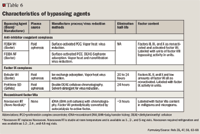
AVAILABLE FACTOR CONCENTRATES

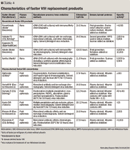
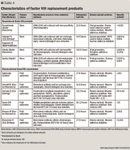



PURITY
The purity of a factor product is the amount or percentage of factor in the total protein content. Purity is generally classified as low (<10 IU/mg protein), intermediate (10 to 100 IU/mg protein), high (100 to 1,000 IU/mg protein), or very high (>1,000 IU/mg protein).13,53 Purity can be affected by the presence of other factors or proteins in the final product, including the addition of albumin. Because of the manufacturing process used, recombinant factor products are high or very high purity.47 Plasma-derived products that undergo monoclonal or affinity chromatography for purification are also of high or very high purity. Plasma-derived products that are low or intermediate purity products are classified as such primarily due to the presence of other proteins, von Willebrand factor, for example, which is the natural carrier of factor VIII.39,41,42
VIRUS REDUCTION METHODS/ISSUES OF SAFETY
Safety of plasma-derived products is especially important to those with hemophilia, because treatment with factor products is life-long. In the early to mid-1980s, a large percentage of patients with hemophilia worldwide were exposed to the human immunodeficiency virus and hepatitis C virus through contaminated plasma-derived factor products.54–57 The incident prompted changes in the collection process for blood and plasma and also led to a requirement that virus reduction methods were to be used during the production of plasma-derived factor products. Available virus reduction methods inactivate or remove any potential viruses from the factor product through treatment with solvent/detergent or with pasteurization, vapor-heat, or terminal dry heat. Nanofiltration is also used to effectively remove viruses by size. Most available products are treated with 2 virus reduction methods to enhance efficacy in eliminating any potential viruses.53
INHIBITOR DEVELOPMENT AND TREATMENT
One of the most important complications of hemophilia treatment is the development of antibodies, or inhibitors, to the exogenous factor.58 The presence of inhibitors does not result in more frequent or severe bleeding; however, inhibitors do make treatment of a bleeding episode more difficult because the efficacy of the factor is decreased. Inhibitor development is much more common in hemophilia A than in hemophilia B, and is reported in up to 38% of patients with hemophilia A.59,60 Factors that may increase the risk of inhibitor development include severity of hemophilia, the type of genetic mutation, family history, and age at diagnosis and first treatment.59 The risk of inhibitor development appears highest during the first 50 days of exposure to factor concentrate.60 The factor product used (recombinant vs plasma-derived) or switching between factor products does not appear to influence inhibitor development.61
HIGH VERSUS LOW INHIBITOR RESPONDERS
Determination of inhibitor titers is important when treating a patient with hemophilia and inhibitors.58,62 Inhibitor titers are measured in Bethesda units (BU). Low inhibitor titers are generally defined as <5 BU/mL. High titers are 5 BU/mL or greater. Patients are considered low responders when exposure to a factor product results in inhibitor titers of <5 BU/mL. In high responders, inhibitor titers may initially be low, but an anamnestic response may occur, rapidly increasing titers to well over 5 BU/mL after exposure to factor concentrate.58
HIGH-DOSE FACTOR CONCENTRATES
Treatment of a bleeding episode in a patient with inhibitors depends on whether the patient is a low or high responder.58,62 For low responders, high-dose factor concentrate may be effective for treatment. Factor VIII dosing of 25 to 40 IU/kg per BU/mL or 40 IU/kg plus an additional 20 IU/kg per BU/mL has been suggested.29,58
For high responders, high doses of factor concentrate may be ineffective.60 However, for life-threatening bleeding in the presence of initially low inhibitor titers, factor concentrate may be tried because increases in inhibitor titers may not be seen for several days following factor exposure.29,58 When factor concentrates do not result in cessation of bleeding or cannot be used, alternative, or bypassing agents are needed.
BYPASSING AGENTS

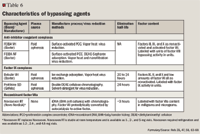
Recombinant factor VIIa is a newer option for treatment of patients with inhibitors. It contains only factor VIIa, therefore, antibody production to factors VIII or IX is not stimulated.60 For this reason, some clinicians prefer recombinant factor VIIa to the plasma-derived bypassing agents.62 Recombinant factor VIIa has a short half-life and more frequent dosing is needed. A dose of 120 μg/kg every 2 hours for severe bleeding has been used, with an increase in dose of up to 300 μg/kg suggested for patients who do not respond to the 120 μg/kg dose.
IMMUNE TOLERANCE THERAPY
Once bleeding has been controlled, long-term eradication of inhibitors, or immune tolerance induction, is frequently considered.62,70 Eradication may be accomplished with several methods and usually involves administration of high-dose factor concentrates for prolonged periods of time. Immune tolerance induction can be used in patients with recent-onset inhibitors or in those with a poor response to bypassing agents, and is more effective in patients with hemophilia A than with hemophilia B (80% vs 30% success rate).60
COMPLIANCE AND COST



Cost may also be a concern for patients with hemophilia as well as for healthcare providers. For recombinant factor VIII concentrates, the average wholesale cost per unit of factor ranges from $1.56 to $1.68.72 For a 50-kg patient who requires an increase in factor VIII levels to 60% of normal, 1,500 units of factor would be needed as an initial dose, with a 50% maintenance dose given every 12 hours. If a 5-day treatment course were needed, the cost could range between $11,700 to $12,600. Although plasma-derived products are less expensive per unit ($1.00 to $1.09 for monoclonal purified products), recombinant DNA-derived products are preferred.73
SUMMARY
Hemophilia is a congenital deficiency of either clotting factor VIII or IX, potentially resulting in spontaneous and life-threatening bleeding. Treatment of hemophilia involves replacement of the deficient factor and needs to be initiated promptly to help reduce long-term complications of the disorder. Patient education is also key in the treatment of hemophilia, not only to ensure that patients can recognize bleeding episodes quickly, but also to allow for a higher quality of life through an understanding of the disorder and the need for comprehensive care.
Dr Stachnik is clinical assistant professor, department of pharmacy practice, College of Pharmacy, University of Illinois at Chicago.
Disclosure Information: The author reports no financial disclosures as related to products discussed in this article.
REFERENCES
1. Colman R, Clowes A, George J, Goldhaber S, Marder V. Overview of hemostasis. In: Colman R, Marder V, Clowes A, George J, Goldhaber S, eds. Hemostasis and Thrombosis. 5th ed. New York, NY: Lippincott Williams & Wilkins; 2006:3–16.
2. Camire R, Pollak E. Genetics of coagulation. In: Colman R, Marder V, Clowes A, George J, Goldhaber S, eds. Hemostasis and Thrombosis. 5th ed. New York, NY: Lippincott Williams & Wilkins; 2006:59–89.
3. Bolton-Maggs P, Pasi K. Haemophilia A and B. Lancet. 2003;361:1801–1809.
4. Norris LA. Blood coagulation. Best Pract Res Clin Obstet Gynaecol. 2003;17:369–383.
5. Colman R, Marder V, Clowes A. Overview of coagulation, fibrinolysis, and their regulation. In: Colman R, Marder V, Clowes A, George J, Goldhaber S, eds. Hemostasis and Thrombosis. 5th ed. New York, NY: Lippincott Williams & Wilkins; 2006:17–20.
6. Knokle B. Bleeding and thrombosis. In: Fauci A, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, Loscalzo J, eds. Harrison's Principles of Internal Medicine. 17th ed. New York, NY: McGraw Hill Medical; 2008:363–369.
7. Wagenman B, Townsend K, Mathew P, Crookston K. The laboratory approach to inherited and acquired coagulation factor deficiencies. Clin Lab Med. 2009;29:229–252.
8. World Federation of Hemophilia. Annual Global Survey 2008. Available at: http://www.wfh.org/2/docs/Publications/Statistics/2008_Global_Survey_Report.pdf. December 2009. Accessed June 15, 2010.
9. Soucie J, Evatt B, Jackson D; Hemophilia Surveillance System Project Investigators. Occurrence of hemophilia in the United States. Am J Hematol. 1998;59:288–294.
10. Bowen D. Haemophilia A and haemophilia B: molecular insights. J Clin Pathol: Mol Pathol. 2002;55:127–144.
11. Kessler C, Mariani G. Clinical manifestations and therapy of the hemophilias. In: Colman R, Marder V, Clowes A, George J, Goldhaber S, eds. Hemostasis and Thrombosis. 5th ed. New York, NY: Lippincott Williams & Wilkins; 2006:887–904.
12. Peyvandi F, Jayandharan G, Chandy M, et al. Genetic diagnosis of haemophilia and other inherited bleeding disorders. Haemophilia. 2006;12(suppl 3):82–89.
13. World Federation of Hemophilia. Guidelines for the management of hemophilia. Available at: http://www.wfh.org/2/docs/Publications/Diagnosis_and_Treatment/Guidelines_Mng_Hemophilia.pdf. 2005. Accessed June 15, 2010.
14. Konkle B. Clinical approach to the bleeding patient. In: Colman R, Marder V, Clowes A, George J, Goldhaber S, eds. Hemostasis and Thrombosis. 5th ed. New York, NY: Lippincott Williams & Wilkins; 2006:1147–1158.
15. Kulkarni R, Soucie J, Lusher J. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–1290.
16. Rodriguez-Merchan E. Articular bleeding (hemarthrosis) in hemophilia. Treatment of hemophilia. 2008;23:1–11. Available at: http://www.wfh.org/2/docs/Publications/Musculoskeletal_Physiotherapy/TOH-23-Hermarthrosis-Revised2008.pdf. Accessed June 15, 2010.
17. Jansen N, Roosendaal G, Lafeber F. Understanding haemophilic arthropathy: an exploration of current open issues. Br J Haematol. 2008;143:632–640.
18. Mannuccci P. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Treatment of Hemophilia. 1998;11:1–11. Available at: http://www.wfh.org/2/docs/Publications/Treatment_Products/Monographs/TOH-11_English_DDAVP.pdf. Accessed June 15, 2010.
19. Stimate [package insert]. King of Prussia, PA: CSL Behring, LLC; 2007.
20. DDAVP Injection [package insert]. Bridgewater, NJ: Sanofi-aventis US, LLC; 2007.
21. DDAVP Nasal Spray [package insert]. Bridgewater, NJ: Sanofi-aventis US, LLC; 2007.
22. DDAVP Rhinal Tube [package insert]. Bridgewater, NJ: Sanofi-aventis US, LLC; 2007.
23. DDAVP Tablets [package insert]. Bridgewater, NJ: Sanofi-aventis US, LLC; 2007.
24. Leissinger C, Becton D, Cornell C. Gill J. High-dose DDAVP intranasal spray (Stimate) for the prevention and treatment of bleeding in patients with mild haemophilia, mild or moderate type I von Willebrand disease and symptomatic carriers of haemophilia A. Haemophilia. 2001;7:258–266.
25. Cyklokapron [package insert]. New York, NY: Pharmacia & Upjohn Company; 2008.
26. Amicar [package insert]. Newport, KY: Xanodyne Pharmaceuticals, Inc.; 2008.
27. Lysteda [package insert]. Newport, KY: Xanodyne Pharmaceuticals, Inc.; 2009.
28. Tengborn L. Fibrinolytic inhibitors in the management of bleeding disorders. Treatment of Hemophilia. 2007;42:1–15. Available at: http://www.wfh.org/2/docs/Publications/Treatment_Products/Monographs/TOH42-Fibrinolytic_Inhibitors.pdf. Accessed June 15, 2010.
29. Shord S, Lindley C. Coagulation products and their uses. Am J Health Syst Pharm. 2000;57:1403–1420.
30. BeneFIX [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc; 2009.
31. Martinowitz U, Luboshitz J, Bashari D, et al. Stability, efficacy, and safety of continuously infused sucrose-formulated recombinant factor VIII (rFVIII-FS) during surgery in patients with severe haemophilia. Haemophilia. 2009;15:676–685.
32. Batorova A, Martinowitz U. Intermittent injections vs. continuous infusion of Factor VIII in haemophilia patients undergoing major surgery. Br J Haematol. 2000;110:715–720.
33. Bidlingmaier C, Deml M, Kurnik K. Continuous infusion of factor concentrates in children with haemophilia A in comparison with bolus injections. Haemophilia. 2006;12:212–217.
34. Recombinate [package insert]. Westlake Village, CA: Baxter Healthcare Corporation; 2006.
35. Helixate FS [package insert]. Tarrytown, NY: Bayer Healthcare LLC; 2009.
36. Kogenate FS [package insert]. Tarrytown, NY: Bayer Healthcare LLC; 2009.
37. Advate [package insert]. Westlake Village, CA: Baxter Healthcare Corporation; 2009.
38. Xyntha [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc; 2008.
39. Alphanate [package insert]. Los Angeles, CA: Grifols Biologicals, Inc.; 2007.
40 Hemofil M [package insert]. Westlake Village, CA: Baxter Healthcare Corporation; 2009.
41. Humate P [package insert]. Kankakee, IL: CSL Behring LLC; 2007.
42. Koate DVI [package insert]. Research Triangle Park, NC: Talecris Biotherapeutics, Inc; 2006.
43. Monarc-M [package insert]. Westlake Village, CA: Baxter Healthcare Corporation; 2005.
44. Monoclate P [package insert]. Kankakee, IL: CSL Behring LLC; 2007.
45. Alphanine SD [package insert]. Los Angeles, CA: Grifols Biologicals Inc; 2008.
46. Mononine [package insert]. Kankakee, IL: CSL Behring LLC; 2007.
47. Brooker M. Registry of clotting factor concentrates. Facts and Figures. 2008;6:1–16. Available at: http://www.wfh.org/2/docs/Publications/Treatment_Products/Monographs/FF6_Registry_8th_2008.pdf. Accessed May 25, 2010.
48. Shapiro A. Anti-hemophilic factor (recombinant), plasma/albumin-free method (octocog-alpha; ADVATE) in the management of hemophilia A. Vasc Health Risk Manag. 2007;3:555–565.
49. Toole J, Pittman D, Orr E, Murtha P, Wasley L, Kaufman R. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci. 1986;83:5939–5942.
50. Gruppo R, Brown D, Wilkes M, Navickis R. Comparative effectiveness of full-length and B-domain deleted factor VIII for prophylaxis-a meta-analysis. Haemophilia. 2003;9:251–260.
51. Kessler C, Gill J, White C, et al. B-domain deleted recombinant factor VIII preparations are bioequivalent to a monoclonal antibody purified plasma-derived factor VIII concentrate: a randomized, three-way crossover study. Haemophilia. 2005;11:84–91.
52. Gruppo R, Brown D, Wilkes M, Navickis R. Increased breakthrough bleeding during prophylaxis with B-domain deleted factor VIII-a robust meta-analytic finding. Haemophilia. 2004;10:449–451.
53. Farrugia A. Guide for the assessment of clotting factor concentrates. 2nd ed. 2008. World Federation of Hemophilia. Available at: http://www.wfh.org/2/docs/Publications/Safety_and_Supply/Regulatory_Guide_2008.pdf. Accessed June 15, 2010.
54. Ragni M, Winkelstein A, Kingsley L, Spero J, Lewis J. 1986 update of HIV seroprevalence, seroconversion, AIDS incidence, and immunologic correlates of HIV infection in patients with hemophilia A and B. Blood. 1987;70:786–790.
55. Ragni M, Tegtmeier G, Levy J, et al. AIDS retrovirus antibodies in hemophiliacs treated with factor VIII or factor IX concentrates, cryoprecipitate, or fresh frozen plasma: prevalence, seroconversion rate, and clinical correlations. Blood. 1986;67:592–595.
56. Goedert J, Brown D, Hoots K, Sherman K. Human immunodeficiency and hepatitis virus infections and their associated conditions and treatments among people with haemophilia. Haemophilia. 2004;10(suppl 4):205–210.
57. Angelotta C, McKoy J, Fisher M, et al. Legal, financial, and public health consequences of transfusion-transmitted hepatitis C virus in persons with haemophilia. Vox Sang. 2007;93:159–165.
58. Haya S, Moret A, Cid A, et al. Inhibitors in haemophilia A: current management and open issues. Haemophilia. 2007;13(suppl 5): 52–60.
59. Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–435.
60. DiMichele D. Inhibitors in hemophilia: a primer. Treatment of Hemophilia. 2008;7: 1–4. Available at: http://www.wfh.org/2/docs/Publications/Inhibitors/TOH-7%20Inhibitor-Primer-Revised2008.pdf. Accessed June 15, 2010.
61. Gouw S, van der Bom J, van den Berg H; CANAL study group. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648–4654.
62. Kempton C, White G. How we treat a hemophilia A patient with factor VIII inhibitors. Blood. 2009;113:11–17.
63. FEIBA NF [package insert]. Westlake Village, CA: Baxter Healthcare; 2009.
64. FEIBA VH [package insert]. Westlake Village, CA: Baxter Healthcare; 2000.
65. Bebulin VH [package insert]. Westlake Village, CA: Baxter Healthcare; 2006.
66. Profilnine SD [package insert]. Los Angeles, CA: Grifols Biologicals; 2008.
67. Novoseven [package insert]. Princeton, NJ: Novo Nordisk, Inc.; 2006.
68. Novoseven RT [package insert]. Princeton, NJ: Novo Nordisk, Inc.; 2010.
69. Hay C, Brown S, Collins P, Keeling D, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133:591–605.
70. Batlle J, Lopez M, Brackmann H, et al. Induction of immune tolerance with recombinant factor VIII in haemophilia A patients with inhibitors. Haemophilia. 1999;5:431–435.
71. De Moerloose P, Urbancik W, VanDen Berg H, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14:931–938.
72. Red Book Updates, June 2010. Montvale, NJ: PDR Network, LLC; 2010.
73. National Hemophilia Foundation. MASAC recommendations concerning products licensed for the treatment of hemophilia and other bleeding disorders. MASAC Document #190. Available at: http://www.hemophilia.org/NHFWeb/Resource/StaticPages/menu0/menu5/menu57/masac190.pdf. Accessed June 15, 2010.


Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).
Management of antipsychotic medication polypharmacy
June 13th 2013Within our healthcare-driven society, the increase in the identification and diagnosis of mental illnesses has led to a proportional increase in the prescribing of psychotropic medications. The prevalence of mental illnesses and subsequent treatment approaches may employ monotherapy as first-line treatment, but in many cases the use of combination of therapy can occur, leading to polypharmacy.1 Polypharmacy can be defined in several ways but it generally recognized as the use of multiple medications by one patient and the most common definition is the concurrent use of five more medications. The presence of polyharmacy has the potential to contribute to non-compliance, drug-drug interactions, medication errors, adverse events, or poor quality of life.
Medical innovation improves outcomes
June 12th 2013I have been diagnosed with stage 4 cancer of the pancreas, a disease that’s long been considered not just incurable, but almost impossible to treat-a recalcitrant disease that some practitioners feel has given oncology a bad name. I was told my life would be measured in weeks.

