
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Management of Parkinson disease: Current treatments, recent advances, and future development
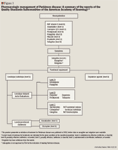
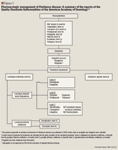
Parkinson disease (PD) is a chronic progressive neurodegenerative disorder that affects >1 million people in the United States. PD causes both motor and nonmotor disturbances; common symptoms include resting tremor, rigidity, and bradykinesia. The current goals of treatment are to slow disease progression and to reduce disability without inducing long-term complications. The classes of agents currently approved for the treatment of PD include levodopa/carbidopa, dopamine agonists, catechol-O-methyl-transferase (COMT) inhibitors, monoamine-oxidase type B (MAO-B) inhibitors, anticholinergics, and agents/ combinations from other drug classes. Newer agents, such as rasagiline and rotigotine, offer additional treatment options to healthcare professionals. Despite these advances in the treatment of PD symptoms, current therapies do not prevent neuron degeneration. Research into new treatments is focused on neuroprotective drugs, new dopamine agonists, and nondopaminergic agents; the goal of these investigative..



Key Points
Abstract
Parkinson disease (PD) is a chronic progressive neurodegenerative disorder that affects >1 million people in the United States. PD causes both motor and nonmotor disturbances; common symptoms include resting tremor, rigidity, and bradykinesia. The current goals of treatment are to slow disease progression and to reduce disability without inducing long-term complications. The classes of agents currently approved for the treatment of PD include levodopa/carbidopa, dopamine agonists, catechol-O-methyl-transferase (COMT) inhibitors, monoamine-oxidase type B (MAO-B) inhibitors, anticholinergics, and agents/ combinations from other drug classes. Newer agents, such as rasagiline and rotigotine, offer additional treatment options to healthcare professionals. Despite these advances in the treatment of PD symptoms, current therapies do not prevent neuron degeneration. Research into new treatments is focused on neuroprotective drugs, new dopamine agonists, and nondopaminergic agents; the goal of these investigative treatments is to slow and ultimately halt disease progression. (Formulary. 2007;42:529–544.)


The loss of dopaminergic neurons leads to both motor and nonmotor symptoms. Cardinal features of PD include resting tremor, rigidity, and bradykinesia. The onset of symptoms is typically asymmetric and commonly starts with resting tremor. Symptoms may become more debilitating over time, with progression to cogwheel rigidity, bradykinesia, and postural instability. Nonmotor symptoms in PD may affect the autonomic, neuropsychiatric, and sensory domains.7 Common neuropsychiatric symptoms include depression, psychosis, and dementia.Common autonomic symptoms include genitourinary dysfunction, gastrointestinal dysfunction, orthostatic hypotension, and thermoregulatory disturbances. Common sensory symptoms include pain, burning, tingling, numbness, and itching.
Unfortunately, there is no cure for PD. Pharmacologic therapy consisting of dopaminergic agents provides symptomatic control for many years; however, the medications used (ie, levodopa) can cause motor complications such as wearing off ("off" time), dyskinesias (drug-induced involuntary movements), and dystonia.8 As the disease advances, treatment becomes more difficult.
TREATMENT





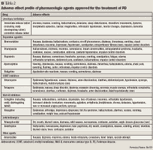
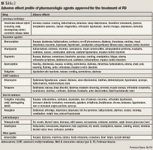
Levodopa/carbidopa. Levodopa is the immediate precursor of dopamine and is converted into dopamine through decarboxylation in the brain and in peripheral tissues. To block some peripheral conversion, thereby preventing nausea and permitting more of the drug to reach the brain, levodopa is combined with carbidopa, a peripheral decarboxylase inhibitor.8 The combination of these 2 drugs is the most effective treatment available for the symptomatic relief of PD. This treatment has demonstrated superior efficacy over dopamine agonists in improving motor function and activities of daily living (ADL) in patients with PD.12 Furthermore, levodopa/carbidopa is possibly neuroprotective for ≥9 months and does not accelerate disease progression (level B). However, there is no evidence to recommend levodopa for long-term neuro-protection (level U).9
This combination therapy is available in both immediate-release (including orally disintegrating tablets) and controlled-release formulations. When initiating levodopa therapy, healthcare professionals may use either product because there is no difference in the rate of motor complications between the 2 formulations (level B).12
Although initially effective for most patients, levodopa/carbidopa treatment will eventually be complicated by periods of inefficiency as the medication wears off and causes dyskinesia.13 Some studies have reported the prevalence of these complications to be as high as 80% in younger patients (onset of PD at age ≤40 years) and 44% in older patients (onset of PD at age >40 years) after 5 years of levodopa treatment.14 As an alternative to administering levodopa more frequently, additional PD medications from different classes can be added to reduce off time. However, with the addition of other medications to increase dopaminergic effects, dyskinesia can be expected to worsen; therefore, dose titration of levodopa/carbidopa is often needed.13
Dopamine agonists. These agents directly stimulate dopamine receptors without the need for metabolic conversion into an active form. Ropinirole and pramipexole are nonergot derivatives that are more selective than other agents in stimulating D2 and D3 dopamine receptors.8 Although levodopa has been demonstrated to be more effective than dopamine agonists (specifically ropinirole and pramipexole) in treating the motor and ADL features of PD, dopamine agonists have the advantage of inducing fewer motor complications such as wearing off, dyskinesias, and on-off motor fluctuations.12 A theoretical explanation for this benefit is a less pulsatile stimulation of dopamine receptors as a result of the longer half-life of the dopamine agonists compared with levodopa.8 Dopamine agonists may also be beneficial as an adjunct to levodopa therapy in patients with advanced PD who experience motor complications. Dopamine agonists do not generate free radicals or induce oxidative stress, thus supporting the theory that they may be more neuroprotective than levodopa. However, there is insufficient evidence to support or refute the use of ropinirole or pramipexole for neuroprotection at this time (level U).9
Neuropsychiatric problems, including hallucinations and psychosis, are more common with dopamine agonists than with other agents used to treat PD, particularly in elderly patients. Impulse control disorders, including compulsive gambling, buying, and sexual behavior, have also been reported with the use of dopamine agonists.15 Cardiac valvular insufficiency is a serious adverse effect that has been reported in patients treated with ergot derivatives and is the reason for the voluntary withdrawal of pergolide from the market by its manufacturers in late March 2007.16
Apomorphine is an injectable nonergot dopamine agonist available for use in advanced disease as a rescue therapy for patients with PD who suffer from episodes of immobility. Apomorphine causes significant emesis; therefore, trimethobenzamide 300 mg TID must be started 3 days before initial apomorphine treatment and continued for ≥2 months.10
Bromocriptine, an ergot-derived dopamine agonist, is used primarily as an adjunct to levodopa/carbidopa therapy because of the high incidence of adverse events (nausea, hallucinations, confusion, and hypotension) and treatment failures associated with bromocriptine monotherapy in patients with newly diagnosed PD.17
COMT inhibitors. Carbidopa will block some peripheral conversion of levodopa to dopamine by inhibiting decarboxylase; however, there is still loss of levodopa in the periphery due to COMT. The COMT inhibitor tolcapone can inhibit both peripheral and, to a lesser extent, central COMT, whereas entacapone, another COMT inhibitor, is only active in the periphery.8 Administration of levodopa with a COMT inhibitor has the potential to provide the brain with a more stable level of levodopa, possibly decreasing the likelihood of patients developing drug-induced motor complications.
The labeling for tolcapone includes a boxed warning regarding serious hepatotoxicity. For patients treated with tolcapone, liver function tests (LFTs) should be obtained at baseline, every 2 to 4 weeks for the first 6 months of therapy, and then periodically as deemed clinically necessary. The drug is to be discontinued if LFTs exceed 2 times the upper limit of normal.18 Tolcapone may also cause explosive diarrhea, which requires drug discontinuation. Entacapone is not associated with liver dysfunction, and no liver function monitoring is required. However, entacapone is associated with both diarrhea and constipation.
MAO-B inhibitors. MAO-B is the predominant enzyme responsible for the breakdown of dopamine in the brain. Selegiline is an irreversible inhibitor of MAO-B. Blocking MAO-B inhibits catabolism and presynaptic reuptake of dopamine, thereby increasing the supply of dopamine available for use in the brain.8 Selegiline monotherapy can be initiated in patients with early-stage PD who do not have functional impairment to provide a mild, symptomatic benefit before the use of levodopa or dopamine agonists (level A).12 One study demonstrated that in patients randomized to selegiline, levodopa therapy could be delayed for 4 months compared with patients treated with placebo.19 The Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism (DATATOP) study demonstrated that in selegiline-treated patients, levodopa therapy could be delayed for approximately 9 months compared with those patients treated with placebo.20 At this time, there are not enough clinical data to confirm neuroprotection as a mechanism for this ability to delay levodopa therapy (level U).9 Researchers have suggested that selegiline's neurotoxic amphetamine metabolites negate any possible neuroprotection conferred by the agent. In patients with PD with motor fluctuations, healthcare professionals may use selegiline to reduce off time (level C).8 Unlike MAO-A inhibitors, which are used to treat depression, MAO-B inhibitors generally do not cause hypertension after ingestion of tyramine-rich foods and therefore can ordinarily be used safely at recommended doses without dietary restrictions.
Anticholinergics. In PD, the decreased activity of dopamine makes the excitatory effects of acetylcholine more pronounced. Anticholinergics can be used to mediate these effects. Clinically, anticholinergics such as trihexyphenidyl and benztropine are used in younger patients with PD (eg, those aged <60 years) in whom resting tremor is the predominant feature. The use of anticholinergics is contraindicated in patients with prostatic hypertrophy or closed-angle glaucoma because of possible aggravation of these disease states. Centrally acting anticholinergics must be used with caution, as they may cause memory impairment, confusion, and hallucinations; these agents, therefore, have a limited role in older patients with PD who have dementia symptoms.8
Other agents. Amantadine is an antiviral medication that antagonizes at N-methyl-D-aspartate (NMDA) receptors. This agent's precise mechanism of action in PD is unknown.8 A neuro-protective effect with amantadine cannot be confirmed at this time (level U).9
RECENT ADVANCES
FDA has recently approved 3 agents (either new molecular entities or new formulations) for the treatment of PD, expanding the pool of pharmacologic agents available to address the diverse elements of this debilitating disease.

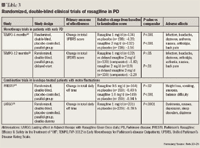
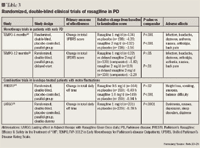
Because sensitivity to tyramine during rasagiline treatment at any dose has not been sufficiently characterized, the labeling for rasagiline includes a warning that patients should avoid tyramine-rich foods, beverages, or dietary supplements and amines (from over-the-counter cough/cold medications) to prevent a possible hypertensive crisis during rasagiline treatment.21
Orally disintegrating selegiline. FDA approved an orally disintegrating formulation of selegiline on June 14, 2006, as an adjunct therapy to reduce off time in patients with PD taking levodopa/ carbidopa. In 140 patients with PD who were taking levodopa and still experiencing ≥3 hours of off time daily, off time was decreased by 32% (2.2 h) in the treatment group versus 9% (0.6 h) in the placebo group (P<.001).27 The Tmax for orally disintegrating selegiline 1.25-mg or 2.5-mg doses is reported to be in the range of 10 to 15 minutes compared with 40 to 90 minutes observed with oral selegiline 5-mg tablets. The Cmax for the orally disintegrating selegiline 2.5-mg dose was 4 times that achieved with selegiline 5-mg tablets administered BID. Because this orally disintegrating formulation bypasses gastric absorption and first-pass metabolism, a lower dose can be used.28 However, the potential clinical benefit of this formulation compared with the conventional selegiline formulation is not clear.
Rotigotine transdermal patch. FDA approved rotigotine, a nonergot dopamine agonist, on May 9, 2007, for the treatment of early PD.29 The safety and efficacy of once-daily rotigotine was assessed in a multicenter, randomized, double-blind study involving a total of 277 patients with early-stage PD. Patients randomized to rotigotine (n=181) received a transdermal patch with an initial dose of 2 mg/24 h, titrated weekly up to 6 mg/24 h for 6 months. The rotigotine-treated patients were reported to have significant improvement in Unified Parkinson's Disease Rating Scale (UPDRS) scores compared with the placebo-treated patients (n=96). UPDRS is a rating scale used to follow the longitudinal course of PD; the scale measures 1) mentation, behavior, and mood; 2) ADL; and 3) motor functions. The rotigotine group had more responders with ≥20% improvement than the placebo group (48% vs 19%, respectively; P<.001). The most commonly reported adverse events were application-site reactions, nausea, somnolence, and dizziness.30 The rotigotine transdermal patch is also being investigated for advanced stages of PD. In the Prospective Randomized Evaluation of a new Formulation: Efficacy of Rotigotine (PREFER) study, which involved 351 patients with advanced PD, a higher dose of rotigotine (8 mg/24 h and 12 mg/24 h) resulted in significant decreases in mean off time (1.8 and 1.2 h/d, respectively) compared with placebo. "On" time without dyskinesia after awakening was more than doubled in both rotigotine treatment groups compared with placebo-treated patients.31
Currently available dopamine agonists require multiple doses daily to attain therapeutic levels. Rotigotine provides convenient once-daily dosing, although the ability of the rotigotine transdermal patch to delay or prevent motor complications has not been investigated. In February 2007, rotigotine was approved in Europe for all stages of PD.32
FUTURE DEVELOPMENT

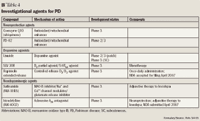
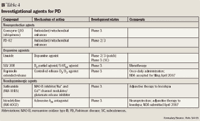
Neuroprotective agents. Agents currently used for the treatment of PD reduce symptoms but become less effective over time. Identifying neuroprotective agents that can slow or stop disease progression will delay the need for dopamine replacement therapy. Only recently have interventional studies been conducted on agents that might slow disease progression. Unfortunately, none of the drugs studied has yet been demonstrated to delay the progression of PD.33 The National Institute of Neurological Disorders and Stroke (NINDS) has organized a nationwide, multicenter effort called Neuroprotection Exploratory Trials in Parkinson's Disease (NET-PD) to study compounds that may slow the clinical decline of PD and that may therefore warrant phase 3 studies to determine potential efficacy.56
Coenzyme Q10 (ubiquinone). In patients with PD, there is a reduction of coenzyme Q10 levels in the mitochondria.34 By enhancing complex I activity in the mitochondrial electron transport, coenzyme Q10 reduces oxidative stress in neurons.35 In a phase 2, placebo-controlled, double-blind clinical study, patients (N=80) were randomly assigned to 1 of 3 dosages of coenzyme Q10 (300, 600, or 1,200 mg/d) or placebo. Patients were followed for progression of disease as measured by UPDRS. Patients treated with the highest dosage of coenzyme Q10 demonstrated a lower rate of disability progression over 16 months compared with the patients who received placebo. This suggests that coenzyme Q10 plays a role in slowing functional decline in patients with PD.36 Another phase 2 clinical study was designed to determine whether further studies might be warranted to evaluate coenzyme Q10's efficacy in controlling the symptoms of early-stage PD. Results demonstrated potential efficacy, thereby suggesting that additional studies might be warranted.37 In a recent trial, coenzyme Q10 100 mg administered TID for 3 months demonstrated no significant symptomatic effects in a group of 131 patients with PD without motor fluctuations.38 This agent is currently in phase 3 trials.
PD-02. PD-02 is an ultrapure clinical form of creatine. As an antioxidant, creatine enhances mitochondrial function and reduces oxidative stress through stabilization of mitochondrial creatine kinase. Creatine may limit apoptosis by inhibiting the activation of mitochondrial transition pore.39 A phase 2 study designed to evaluate the utility of PD-02 demonstrated that the rate of disease progression in patients treated with this agent was lower than predetermined thresholds.40 Patients are currently being enrolled for a phase 3 trial.41








FDA Issues Complete Response Letter for Pz-Cel to Treat Epidermolysis Bullosa
April 22nd 2024Prademagene zamikeracel is a cell therapy designed to incorporate the functional collagen-producing COL7A1 gene into a patient’s own skin cells. The FDA is asking for additional information on manufacturing practices.

