
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Metastatic breast cancer: A review of current and novel pharmacotherapy
Despite advancements, treatment of metastatic breast cancer hinges on multifarious factors and numerous unanswered questions about therapy linger. Agents that are highly active in heavily pretreated patients are needed to optimize outcomes in patients with metastatic disease. This article reviews current and novel treatment options for metastatic breast cancer.



Key Points
Abstract

Despite improvements in screening and treatment, breast cancer remains the most common cause of cancer and the second-leading cause of cancer-related death in women. It is expected that 1 in 8 women will develop breast cancer in their lifetime, and for 2010 it was estimated this would result in 39,000 deaths and over 207,000 newly diagnosed cases.1,2 Most patients present with an early stage of breast cancer, but 6% to 10% of patients initially present with metastatic breast cancer (MBC), defined as cancer occurring at sites distant from the breast, chest wall, and regional lymph nodes (mainly bone, lung, liver, and brain).3 However, most patients who present with MBC have a recurrence of early-stage breast cancer.4,5
Research efforts focusing on tumor biology, specifically cellular signaling pathways, have aided the development of multiple therapeutic options that target cellular proliferation, survival, and angiogenesis. These agents improve outcomes when used alone or in combination with chemotherapy. However, selecting optimal therapy for patients with MBC depends on multifarious factors, and there are numerous unanswered questions about therapy. Although initial therapy may provide good response, disease progression eventually ensues. Most patients progress within 12 to 24 months; therefore, novel treatments that prolong quality and quantity of life are greatly needed.3,6–9 This article reviews both contemporary therapies and novel agents used in MBC.
ENDOCRINE THERAPY FOR MBC

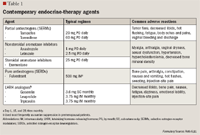
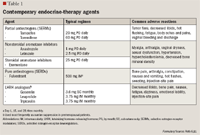
Selective estrogen-receptor modulators. Estrogen, on binding to the nuclear ER-a receptor, has profound proliferatory effects on mammary epithelial cells and is a well-established risk factor for breast cancer development. The ER is overexpressed in as many as 70% of women with breast cancer and is therefore an important target for therapeutic manipulation.4,11–13
For several decades, the SERM tamoxifen (TAM) has been the cornerstone of therapy in ER- and PgR-positive breast cancers.11–13 SERMs work as competitive ER antagonists. Because ERs vary in chemical structure from tissue to tissue, the activity of a SERM is not always antagonistic. Hence, TAM possesses pharmacologic properties that are both beneficial and detrimental in breast cancer patients. TAM exerts agonist activity on the uterine endometrium, the coagulation system, the liver, and bone. Agonist activity is responsible for both desirable (preserved bone mineral density, reduced low-density lipoprotein cholesterol) and unwanted (endometrial hypertrophy, vaginal bleeding, increased endometrial cancer risk, and thromboembolism) effects.4,11–13
TAM plays an important role in the treatment of both premenopausal and postmenopausal women with MBC. In premenopausal patients with MBC, TAM in combination with luteinizing-hormone?releasing hormone analogues (LHRH)/ovarian ablation remains a first-line option.6,7 Although an effective combination, many clinicians prefer not to rely on LHRH suppression, giving preference to oophorectomy. Postmenopausal MBC patients who are antiestrogen naive or who are more than 1 year from prior antiestrogen therapy may also be placed on TAM.3,5–7
An equally effective alternative to TAM is toremifene.6,14 Studies have shown that toremifene is associated with a lower incidence of secondary endometrial cancer and thromboembolic complications as compared with TAM.12,14 Although toremifene is safe and effective, it is mainly used as an alternative to TAM in patients with impaired CYP2D6 metabolism or patients receiving concomitant CYP2D6 inhibitors, as recent data have revealed that on TAM treatment these patients had a greater risk for breast cancer recurrence.15 TAM's antagonist activity is largely a product of CYP2D6-derived metabolites. Toremifene, however, does not require CYP2D6-mediated activation or metabolism and is a viable option, although recent outcome data for this agent are scarce.
Aromatase inhibitors. AIs comprise another class of endocrine agents that are very effective in both premenopausal and postmenopausal women with MBC. The majority of circulating estrogen in postmenopausal women is derived from the peripheral conversion of androgens to estrogens by the enzyme aromatase.3,6,11–13 AIs inhibit the action of the enzyme aromatase, decreasing over 90% of circulating estrogen in postmenopausal women. In premenopausal women, however, the use of AIs alone is not considered effective as ovarian function can enhance the production of aromatase resulting in estrogen expression in peripheral tissues.
In considering the role of AI in MBC, comparing its efficacy as well as toxicity with that of other endocrine agents should be noted. Based on available data, it is reasonable to consider TAM-LHRH/oophorectomy or AI-LHRH/oophorectomy in premenopausal patients.6 In postmenopausal MBC, the Cochrane Collaboration found that AIs were more beneficial than other endocrine therapies because they decreased disease progression and improved survival by 10%, supporting their use in this population.16 Side-effect profiles for endocrine therapies vary, and patient tolerability plays an important role when choosing therapy. Compared with TAM, AI were associated with a higher incidence of nausea and arthralgia/myalgia (affecting 5% to 38% of patients) but a lower incidence of vaginal bleeding, uterine-specific adverse effects, and thromboembolism. Long-term therapy with AIs is associated with hypercholesterolemia, hypertension, and a significant degree of bone-mineral loss resulting in fracture risk.11–13 Vigilant monitoring of lipids, blood pressure, and bone-mineral density is required, as these are predictable and treatable side effects. Based on available data and patient tolerability, AI-LHRH/oophorectomy can be considered first-line therapy, along with TAM-LHRH/oophorectomy, in premenopausal patients and as first-line therapy over TAM in postmenopausal patients with MBC.6,16
Selective estrogen-receptor downregulators. The newest agent for use in hormone-sensitive MBC is fulvestrant (FLV), a steroid-based pure antiestrogen. FLV is a 17 -estradiol analog that binds to the ER, disrupting its expression and ultimately accelerating ER protein degradation. This new class is referred to as "SERDs."17,18 Initial studies that compared FLV, 250-mg intramuscular dose given every 28 days, with TAM and AI in postmenopausal women with MCB found FLV to be no different in time to tumor progression and overall survival.19,20 However, a select group of patients in these early trials receiving FLV had early progression, suggesting the need for dose optimization.19,20 Subsequent phase 2 studies using a 500-mg high-dose regimen were associated with improved outcome demonstrated by decreased risk of disease progression without additional toxicity.21,22 When compared with anastrozole in first-line treatment for advanced breast cancer, high-dose FLV was superior, significantly extending time in remission, equating to longer disease control in patients with advanced disease.21 More recently, investigators have discovered that in a postmenopausal breast cancer model the combination of FLV and letrozole suppresses estrogen-stimulated growth factors better than either agent alone.23
Recently, the CONFIRM study reported that progression-free survival was significantly longer for high-dose FLV compared with the 250-mg dose of FLV in recurrent MBC (median progession-free survival, 6.5 vs 5.5 months; HR, 0.80; 95% CI, 0.68–0.94; P=.006), corresponding to a 20% decrease in progression risk.22 Because of the positive outcomes in MBC with high-dose FLV over the 250-mg dose, FDA recently approved high-dose FLV for hormone-receptor-positive, postmenopausal MBC, as two 250-mg intramuscular injections, 1 in each buttock on days 1, 15, and 29, and monthly thereafter. Consideration is now being given to future studies using larger FLV doses in MBC that is poorly responsive to endocrine therapy. FLV is well tolerated, with the most common side effects being mild to moderate bone pain, arthralgia, constipation, nausea and vomiting, hot flashes, sweating, mild liver enzyme elevations, and pain at the injection site.18–22 Although more studies with this agent are needed, considering the recent data from CONFIRM, FLV's efficacy in MBC and increased duration of response indicates the need to reassess its place in the endocrine therapy hierarchy.
In hormone-sensitive disease, first-line hormone therapy is associated with an average of 8 to 12 months of response, while subsequent therapy, initiated at the time of progression, is associated with an additional 4 to 6 months of tumor control.11–14 This being the case, the best regimen in terms of dosage, combination of drugs, and sequence of administration remains to be fully elucidated. Ongoing and future clinical trials will hopefully clarify this situation.
CHEMOTHERAPY FOR MBC
In MBC, chemotherapy usually is reserved for patients who are hormone resistant, in visceral crisis, or who have rapidly progressive disease. Chemotherapy is given as monotherapy, combination therapy, or as a single-agent sequential regimen.3–7 The National Comprehensive Cancer Network (NCCN) guidelines recommend several preferred single-agent and combination chemotherapy regimens in MBC.6,7 The practice guidelines give preference to single-drug sequential therapy for the majority of patients; however, a significant degree of controversy exists regarding the optimal agent, dosage, sequence, and duration of therapy. Of paramount importance in chemotherapeutics is careful consideration of the efficacy-to-toxicity ratio. Although combination therapy is rapidly more active, with a higher objective response and longer time to tumor progression, increased toxicities and frequently reported lack of improvement in overall survival limit its utility, especially in a patient population where treatment is aimed at maintaining and improving quality of life.3-7 Anthracycline (largely doxorubicin) and taxane (docetaxel and paclitaxel) -based regimens are highly active in MBC.6,7,24–26 Unfortunately, because anthracyclines and taxanes are standard of care in the nonmetastatic adjuvant setting, their role in MBC is limited due to resistance and cumulative toxicity issues.



NOVEL CHEMOTHERAPEUTIC AGENTS
Capecitabine. Capecitabine (CAP), an oral prodrug of 5-fluorouracil, has been successfully used as mono-, sequential, and combination therapy in anthracycline- and taxane-pretreated patients with MBC. In phase 2 monotherapy trials, CAP produced an average response of 22%, median time to progression of 3.7 months, and median overall survival ranging from 10.1 to 15.2 months.27–29
Recent data from MONICA, a nonrandomized, prospective, phase 2 study, showed that monotherapy with 2,000 mg/m2 of CAP was safe and effective when used first-line in HER2-negative MBC. In this study, median time to tumor progression and overall survival were 7.3 and 17.1 months, respectively.30 In patients with MBC progressing after anthracycline and taxane therapy, the combination of CAP and ixabepilone has been shown to increase median survival and overall survival as compared with CAP alone.31 When taking into account efficacy and tolerability, CAP dosing must be considered. When CAP is used as mono- or sequential therapy, a dose of 1,250 mg/m2 twice daily (2,500 mg/m2 daily) for 14 days every 21 days has been shown effective and well tolerated.27–29 However, when combined with other chemotherapeutic agents, added bone marrow toxicity is of concern and daily doses of 2,000 mg/m2 are preferred.29,31,32 Finally, CAP in combination with growth-factor-targeting agents is also associated with positive outcomes in MBC.
In the clinical setting, CAP is effective and, overall, well tolerated. Side effects most frequently associated with CAP are fatigue, diarrhea, peripheral neuropathy (including hand-foot syndrome), anemia, thrombocytopenia, and nausea. The majority of these side effects are mild to moderate (grade 1-2) and generally treatable or reversible on therapy discontinuation. However, cases of severe (grade 3-4) neuropathy (hand-foot syndrome), diarrhea, and bone marrow suppression have occurred.27–32 Despite these toxicities, CAP is an effective therapy in MBC as mono-, sequential, and combination therapy. Its availability as an oral therapy provides patients with a convenient and effective treatment option, but this must be weighed against the potential for compliance issues that might adversely affect therapeutic outcomes.
Gemcitabine. As a nucleoside pyrimidine analogue, gemcitabine's antitumor activity is attributed to inhibition of DNA polymerization and RNA synthesis. In addition to its use as monotherapy, gemcitabine has been used in combination with anthracyclines, taxanes, cisplatin, and growth factor- and angiogenesis-targeting agents.32–36 As first-line therapy in MBC, Blackstein et al reported an overall response rate (ORR) of 37.1% and a median time to tumor progression of 5.1 months, which was coupled with favorable tolerability, grade 4 toxicities being rarely reported. However, grade 3 neutropenia, thrombocytopenia, and nausea and vomiting occurred in 30.3%, 6.3%, and 10.3% of patients, respectively.35 In anthracycline- and taxane-pretreated patients, gemcitabine dosed at 800 mg/m2 on day 1, 8, and 15 of a 28-day cycle was associated with ORR of 17% and a median survival of 9.5 months.36 Significant grade 3 toxicities were rare but included neutropenia (14%), thrombocytopenia (9%), anemia (5%), elevated liver function tests, constipation, and fatigue.
These data support gemcitabine as an attractive and well-tolerated agent in MBC. Given its distinct mechanism of action and lack of overlapping toxicity with prior treatment exposure, this agent represents an important option in pretreated patients who have experienced treatment-limiting toxicities. The NCCN guidelines recommend it as a preferred agent in combination with paclitaxel at a dose of 1,250 mg/m2 and as a preferred single agent at a dose ranging from 800 to 1,200 mg/m2 . Although not highly active in heavily pretreated patients (those exposed to 3 or more chemotherapy regimens), this agent is a reasonable option, especially in elderly patients, where it is both active and well tolerated.6
Nanoparticle albumin-bound (nab)-paclitaxel. Taxanes are highly active in breast cancer; however, their utility is limited by acute side effects (mainly hypersensitivity reactions), cumulative toxicity issues (most commonly neuropathy), and acquired tumor resistance.37–39 Both paclitaxel and docetaxel are formulated with complex solvent systems.38–39 These solvents are biologically and pharmacologically active and are responsible for many of the toxicity issues that limit utility and dose escalation. Cremophor EL, the vehicle in solvent-based paclitaxel, is believed to contribute to paclitaxel's hypersensitivity reactions, neuropathy, and myelosuppression.39Nab-paclitaxel was developed to avoid the acute toxicities and dose-limiting side effects associated with solvent-based paclitaxel.39 The result is a 120- to 150-nm nanoparticle colloidal paclitaxel suspension. Compared with solvent-based pac-litaxel, nab-paclitaxel can be given in a shorter infusion time, without the use of hypersensitivity-blocking premedications. In addition, the nab formulation enhances tumor drug delivery, allowing for higher dose-intensity, which ultimately leads to enhanced antitumor activity.39 Preclinical data showed that in addition to improved tolerability, nab-pac-litaxel uptake in tumor tissue was greater than that observed with solvent-based paclitaxel.37–39 Compared with solvent-based paclitaxel (175 mg/m2 over 3 hours), nab-paclitaxel (260 mg/m2 over 30 minutes) given every 3 weeks was associated with a significantly higher ORR (33% vs 19%) and longer time to tumor progression (23 vs 16.9 weeks).40 Additionally, overall side effects were not significantly different between treatment groups. Sensory neuropathy was significantly more common in nab-paclitaxel-treated patients, but the duration of neuropathy was much shorter and did not negatively impact quality of life.
A subsequent study reported by Gradishar et al compared nab-paclitaxel to docetaxel as first-line therapy for MBC.41 Weekly nab-paclitaxel at 150 mg/m2 was associated with significantly longer progression-free survival (12.9 vs 7.5 months), and greater ORR (74% vs 39%) compared to docetaxel. Furthermore, weekly nab-paclitaxel has been shown to be effective and well tolerated in MBC patients heavily pretreated with taxanes.41,42
With regard to safety, the most common treatment-associated adverse effects include alopecia, neutropenia, fatigue, arthralgia, and sensory neuropathy, none of which are greater in occurrence or severity compared to that of solvent-based taxanes.40–43 Because nab-paclitaxel has increased efficacy without increased toxicity in addition to preserved activity in heavily pretreated taxane patients, it is considered an advantageous alternative in contemporary MBC management and is listed as a preferred single agent in the practice guidelines.6
Ixabepilone. Epothilones are a new class of antineoplastics, derived from the myxobacterium Sorangium cellulosum, that stabilize microtubule dynamics, halting mitosis and leading to apoptotic cell death.43-45 Although mechanistically similar to taxanes, preclinical data have demonstrated this agent is not affected by current mechanisms linked to taxane resistance. A great advantage of this new class is activity in taxane-resistant and taxane-insensitive tumor cell lines.43,44
Ixabepilone (IXA) is the first epothilone to gain FDA approval when used in combination with CAP for patients with MBC who have failed anthracycline and taxane regimens and as monotherapy after CAP failure. This semisynthetic analog of epothilone B has 2 to 10 times greater tubulin-polymerizing activity than paclitaxel, is not affected by current mechanisms of taxane resistance, and remains active in taxane-resistant cell lines. In addition, unlike paclitaxel, IXA exhibits linear pharmacokinetics permitting better estimations of dose-related toxicity.44,45 In phase 2 studies, IXA monotherapy was associated with an ORR of up to 12% in patients considered heavily pretreated or taxane resistant, while ORR of up to 57% have been observed in taxane-naive patients.46–49 Phase 3 studies have evaluated the combination of IXA/CAP in patients with anthracycline and taxane resistance.31,50 The combination of IXA/CAP significantly improved progression-free survival and response rate compared with CAP alone, but a significant difference in overall survival was not noted. Despite lack of a significant impact on overall survival, an observed difference in median overall survival (16%) favored the IXA-containing treatment arm. Additionally, preclinical studies showed IXA has synergy when combined with trastuzumab (TRAS) and bevacizumab (BVB). These combinations are discussed below.
While effective, grade 3-4 neurotoxicity is a common occurrence with this agent. To combat neurotoxicity, alternate dosing schedules have been evaluated but have not demonstrated improved response or decreased toxicity.46–50 Common side effects observed in the clinical setting include neutropenia, peripheral neuropathy (in as many as 60% of patients), fatigue, myalgia/arthralgia, alopecia, and nausea and diarrhea. When combined with CAP, grade 3-4 neutropenia and neuropathy (frequently requiring intervention) occur at a significantly higher rate compared with monotherapy. Despite associated toxicity, further analysis of study data has concluded that a positive risk/benefit ratio supports the use of IXA/CAP over CAP alone in progressive disease.51,52 Taken together, the combination of IXA/CAP is effective in patients with limited treatment options and in patients who progress on CAP and/or taxane-based regimens. As anthracycline/taxane combination therapy is a standard-of-care approach in invasive breast cancer, IXA alone or in combination offers an important therapeutic option in MBC, especially in the setting where tumors have acquired taxane resistance.
Eribulin. A recent addition to the MBC treatment regimen is eribulin, a synthetic analog of halichondrin B originally isolated from the marine sponge Halichondria okadai. 53,54 This non-taxane microtubule dynamics inhibitor has antitumor efficacy with acceptable tolerability and is the only single agent that has prolonged overall survival in heavily pretreated MBC patients.53,54 Eribulin's mechanism of action is unique in that it blocks polymerization of tubulin but does not affect microtubule depolymerization. In addition, eribulin depletes nuclear tubulin stores, the combined effect driving cell arrest and apoptosis. Phase 3 study results from the EMBRACE trial, which compared eribulin to a treatment of the physician's choice (96.4% chemotherapy regimens), showed that in heavily pretreated MBC patients (2 to 5 prior chemotherapies that included an anthracycline and a taxane), eribulin significantly positively affected the primary end point of overall survival by a median of 2.5 months (P=.04) while increasing objective response and clinical benefit rates.55
Eribulin was well tolerated; the incidence of serious side effects were comparable between treatment arms. The most common reported side effects, fatigue and peripheral neuropathy, were low in incidence and rarely greater than grade 3. Neutropenia, another commonly reported side effect, was grade 4 in 24.1% of patients although febrile neutropenia was rare (4.6%). Eribulin's water solubility obviates the need for solvent vehicles, allowing for decreased delivery time and avoiding hypersensitivity reactions and hypersensitivity premedication administration.53,54 It is expected that in future and ongoing trials, the combination of eribulin with other chemotherapeutic and biology targeting agents will better position this agent for use in MBC. Eribulin's efficacy combined with an acceptable degree of tolerability makes this an attractive new agent.
TUMOR-BIOLOGY-TARGETING AGENTS
Recent advances in our understanding of tumor biology and the subsequent discovery of several signal-transduction pathways have increased our awareness of factors involved in tumorigenesis, tumor growth, chemoresistance, and metastatic spread. Today, biological agents directed at molecular targets are finding their place in modern cancer regimens.5–7
Epidermal growth factor receptors (EGFR) are promising molecular targets in MBC. EGFR is a membrane-bound tyrosine kinase 1 receptor involved in a variety of signal transduction pathways associated with carcinogenesis of tumor cells. EGFR belongs to the ErbB family, of which there are currently 4 known members: Erb1 (EGFR), ErbB2 (HER2/neu), ErbB3, and ErbB4.56–58 As stated earlier, HER2 overexpression is associated with increased recurrence, tumor invasiveness, decreased overall survival, and poor chemotherapy responsiveness and as such represents an important target for strategic biologic therapy.9,10
Another modern cancer target is angiogenesis. To date, 3 vascular endothelial growth factor receptors (VEGFR 1 to 3) that bind vascular endothelial growth factor (VEGF) have been identified. VEGF expression is upregulated in tumor cells and is responsible for vascular migration, invasion, and ultimately the establishment of a blood vessel network essential for distant metastatic proliferation. High VEGF tumor expression is associated with metastasis, early relapse, and decreased survival, so anti-angiogenesis represents another important biologic target in MBC.4,8 In addition to the targets mentioned above, a variety of new growth factor receptors, intracellular signal transduction pathways, and DNA-level enzymatic pathways are being discovered. Their role in carcinogenesis is being appraised, and they may offer other ideal targets for future drug development.4,10,56,59
Trastuzumab. TRAS, a humanized monoclonal antibody, was the first agent developed to target HER2 overexpression. This agent is known to exert antitumor effects through multiple proposed mechanisms that include activation of natural killer cells through antibody-dependent cellular cytotoxicity, inhibition of HER2 signal transduction leading to cell cycle arrest, inhibition of angiogenic factor release and/or expression, and inhibition of DNA repair gene transcription leading to cellular DNA damage.56–58
TRAS represents an effective therapy in patients with MBC who overexpress HER2. The Multinational Study in HER2-Overexpressing Metastatic Breast Cancer found that TRAS was associated with a 9.1-month median duration of response and 13-month median duration of survival.60 TRAS was well tolerated; the most clinically significant adverse event, cardiac dysfunction, occurred in 4.7% of patients. When combined with chemotherapy in first-line intervention studies, TRAS was associated with a significantly higher ORR, prolonged progression-free survival, and increased overall survival compared with chemotherapy alone.61 In combination with taxane therapy, TRAS improved time to tumor progression and increased overall survival as compared with taxane therapy alone.61 Preliminary data comparing vinorelbine/TRAS to docetaxel/TRAS in HER2-positive MBC showed a significant difference in time to tumor progression, favoring the vinorelbine/TRAS combination. However, median overall survival of 39 versus 36 months was not significantly different between respective groups.62 The study also concluded that the vinorelbine/TRAS combination was better tolerated as noted by less grade 3-4 leukopenia, febrile neutropenia, and neuropathy.
In anthracycline-pretreated patients with MBC, the combination of TRAS/CAP/docetaxel was associated with a median overall survival of 25.5 months, offering a significant survival advantage and favorable safety profile.63 In the first-line setting, the combination of TRAS/epirubicin/cyclophosphamide (the HEC regimen) was associated with better tumor response and increased time to tumor progression as compared with the EC regimen, which lacked TRAS. In HER2-positive patients, HEC-60 (60 mg/m2 epirubicin) was not only better tolerated, it was associated with greater efficacy outcomes than those of the HEC-90 (90 mg/m2 epirubicin) regimen. Although dose-limiting cardiotoxicity was higher in the TRAS-containing arm, these events were manageable and not associated with cardiac-related deaths.64
Despite its efficacy, TRAS is associated with several serious adverse reactions, including cardiotoxicity, pulmonary toxicity, and infusion-related reactions.60–65 Patients with preexisting cardiac dysfunction, or those exposed to anthracycline or paclitaxel therapy, should only be given TRAS if the benefits clearly outweigh the risk of worsening heart function and possible cardiac events. Assessment of cardiac function is necessary before, during, and after completion of therapy, especially in patients with low baseline ejection fraction or in patients who have been exposed to a high cumulative dose of anthracycline, since the risk for myocardial dysfunction is increased. When used as single or combination therapy, appropriate monitoring for TRAS cardiotoxicity should include baseline evaluation of left ventricular ejection fraction, repeated every 3 months during treatment and then every 6 months at treatment completion for a minimum of 2 years. Pulmonary reactions also are of great concern during TRAS therapy, which necessitates vigilant patient screening for the presence of preexisting pulmonary disease. Pulmonary reactions typically occur within 24 hours of administration and can include dyspnea, acute respiratory distress syndrome, and interstitial pneumonitis. Although not strictly contraindicated, caution should be used in patients with preexisting pulmonary disease. Infusion reactions are also common but are generally mild and typically include fever and chills; however, severe reactions causing hypotension, dyspnea, and rigor have been reported. Pretreatment with acetaminophen and diphenhydramine is usually sufficient to prevent most infusion-related reactions.
TRAS represents a significant new treatment approach to MBC and is recommended by NCCN as a preferred first-line agent for all HER2-positive MBC in combination with paclitaxel (with or without carboplatin), docetaxel, vinorelbine, or CAP.3,5,6 In addition, in patients that progress on TRAS therapy, evidence supports its continued role in slowing further disease progression, decreasing metastatic spread to the central nervous system (CNS), and preventing or delaying chemoresistance.65
Lapatinib. As opposed to targeting the extracellular domain of HER2, inhibition of the intracellular tyrosine kinase domains provides a second effective approach to treating HER2-overexpressing MBC. Like other tyrosine kinase receptor inhibitors (TKI), lapatinib (LAP) competitively inhibits the tyrosine kinase activity of EGFRs, including HER2/HER1. The inhibition of HER2/HER1-mediated signal transduction leads to decreased cellular proliferation and increased apoptosis.4,66–68
Studies using LAP for HER2-positive MBC showed it to be minimally effective when used as a monotherapy; therefore, FDA indications only support combination therapies.69,70 When combined with CAP, significant increases in median time to tumor progression (8.4 vs 4.4 months) and progression-free survival (8.4 vs 4.1 months) were noted, with a trend toward increased overall survival (75.0 vs 64.7 weeks, P=.210) as compared with CAP alone.71,72 CAP-naive patients had significantly longer time to tumor progression (26 vs 15 weeks) compared to patients with prior CAP exposure.68 In addition, the combination of CAP and LAP may have an additional advantage over antibody therapy (eg, TRAS) in patients with CNS metastasis, as it has been observed to prevent CNS metastases, reduce CNS tumor burden, and decrease neurologic side effects.68 The side effect profile of LAP/CAP is likewise favorable, as the combination has been shown only to increase the incidence of diarrhea, rash, and dyspepsia compared with CAP alone.72
LAP also has been studied in combination with letrozole for HER2-positive hormone receptor-positive MBC in postmenopausal women.73 Patients who received LAP plus letrozole had increased progession-free survival (8.2 vs 3 months, P=.008), clinical benefit rate (48% vs 29%, P=.003), and objective response rate (28% vs 15%, P=.021) compared with letrozole alone, although incidences of diarrhea, rash, nausea, and fatigue were significantly higher in the combination group.73
In vitro evaluation of the combination of TRAS/LAP indicated a synergistic effect between these compounds, with LAP inhibiting HER2 downregulation and ubiquitination resulting in an increase in TRAS-mediated cytotoxicity.66,74 Clinically, the combination of LAP 1,000 mg once daily and TRAS 2 mg/kg weekly (following the initial 4 mg/kg loading dose) was compared with LAP 1,500 mg once daily in patients progressing on TRAS-containing regimens.74 The combination resulted in significantly better progression-free survival (12 vs 8.1 weeks), and a trend toward improved overall survival was observed (51.6 vs 39 weeks, P=.106). Diarrhea was the only adverse event observed significantly more often in the combination group (60% vs 48%), while cardiotoxicity (defined as a decrease in LVEF >20% from baseline or below institutional lower limit of normal) was observed in 8 patients in the combination group and 3 in the LAP monotherapy group.
LAP, similar to other TKIs, is associated with serious adverse effects including rash (~30% of patients), hepatotoxicity (black box warning), diarrhea, pulmonary toxicity, and QTc prolongation.67,75,76 A key concern with LAP use is the potential for additive cardiotoxicity. LAP has been associated with asymptomatic, reversible decline in ejection fraction, which may pose problems in patients with prior exposure to anthracyclines, taxanes, or TRAS. Approximately 95% of patients enrolled in trials had previous anthracycline or TRAS exposure. In most reports, however, cardiac dysfunction occurred in only approximately 0.5% of patients and is generally reversible upon discontinuation of therapy.68,71,75 The most commonly reported adverse reaction occurring in 40% to 65% of patients is diarrhea, and it is more common in patients receiving combination therapy with CAP.75,76 Although common, it infrequently requires dose reduction, has rarely led to therapy discontinuation, and is responsive to standard antidiarrheal medications including loperamide and diphenoxylate/atropine.
LAP is recommended for use in HER2-overexpressing MBC when used in combination with CAP for patients who have received prior anthracycline, taxane, and TRAS therapy. It is also approved for use in combination with letrozole in patients with MBC who have disease that is endocrine sensitive and HER2-positive, where the combination has been shown to delay progression and theoretically reestablish or prolong tumor hormone therapy sensitivity.3,6,73 The combination CAP/LAP may be advantageous in patients that intravenous therapy would prove problematic. However, providers must weigh the advantages of an oral chemotherapy regimen with the potential for decreased compliance and ultimately efficacy due to the shift in responsibility of administration to the patient.77
Bevacizumab. Despite the development of HER2-targeted therapies, biotargeting therapy for patients with HER2-negative MBC is currently limited to BVB. BVB is a humanized monoclonal antibody directed against VEGF that has been shown to be beneficial in multiple types of metastatic disease. Targeting VEGF reduces angiogenesis, thereby decreasing tumor growth and permitting a synergy between conventional chemotherapy, ultimately opposing tumor survival.8
BVB was approved by FDA in 2008 as a first-line treatment option in patients with metastatic HER2-negative breast cancer in combination with paclitaxel. This indication was based on results of the E2100 trial, which did not show BVB to increase overall survival but showed BVB to increase median progression-free survival from 5.9 months on paclitaxel alone to 11.8 months when BVB was added to paclitaxel (P<.001).78 The combination produced significant increases in grades 3 or 4 infections (9.3% vs 2.9%), fatigue (8.5% vs 4.9%), hypertension (14.8% vs 0.0%), cerebrovascular ischemia (2% vs 0%), headache (2.2% vs 0%), and proteinuria (3.6% vs 0%) compared to paclitaxel alone.78 An independent, blinded review of the E2100 data supported the results, further validating the role of BVB in HER2-negative disease.79
Subsequently, the RIBBON-1 trial expanded the use of BVB in HER2-negative MBC, using it in combination with either CAP, a taxane, or an anthracycline.80 This trial reported significant increases in progression-free survival when BVB was added to CAP or to an anthracycline- or taxane-based chemotherapy regimen.80 However, these results conflict with a 2005 study that did not show benefit in progression-free survival with the addition of BVB to CAP in heavily treated MBC patients.81
Furthermore, a separate trial in HER2-negative MBC, dubbed the AVADO trial, compared 2 doses of BVB (7.5 mg/kg or 15 mg/kg) in combination with 100 mg/m2 docetaxel to docetaxel monotherapy, each given on day 1 of a 21-day cycle. The 15 mg/kg arm showed a significant increase in progression-free survival compared with docetaxel alone regardless of prior therapy (10.1 versus 8.2 months); however, the 7.5 mg/kg arm only showed a significant increase in progression-free survival when patients were stratified based on prior therapy (9 versus 8.1 months).82 While the 15 mg/kg dose appeared to be more effective, it was associated with 1 grade 5 bleeding event and 1 report of a grade 5 gastrointestinal perforation.82 All-grade bleeding was higher in both the 7.5 mg/kg arm (48.4%) and the 15 mg/kg arm (49.4%) compared with placebo (19.5%).82 A second concern raised by the AVADO trial was the numerically higher overall survival in the placebo group (31.9 months) compared with those of the 7.5 mg/kg arm (30.8 months) and 15 mg/kg arm (30.2 months).82
As side effects appeared to outweigh breast cancer treatment benefits, the FDA advisory committee recommended removing the breast cancer indication from BVB. Subsequent to FDA's initial recommendation, further support for the use of BVB in MBC was provided by Valachis et al and by an open-label study of 2,251 patients.83,84 Both of these studies concluded that the benefits of adding BVB to taxane-based chemotherapy regimens for first-line treatment of MBC outweighed the potential risk of serious adverse events.83,84 These conclusions were further supported by the NCCN, which upheld their treatment recommendation supporting the use of BVB in conjunction with paclitaxel as a preferred treatment regimen for recurrent or metastatic breast cancer.6
Very recently, however, FDA ruled that BVB is associated with an unacceptable efficacy-to-toxicity ratio where toxicity along with a lack of a survival advantage overshadows improvements observed in progression-free survival.85
FDA's decision is not expected to affect BVB availability, nor is it expected to disrupt therapy in patients currently receiving the agent. Furthermore, oncologists are still able to use the agent off-label, but at a cost of $100,000 per year per patient, concerns over reimbursement may curb use of the drug in MBC. It may be that a particular subgroup of patients derives benefit from BVB wherein the benefit outweighs toxicity issues, but identifying this subgroup and evaluation of BVB in this population requires further exploration.
CONCLUSION
Despite few recent advances in endocrine therapy, in the setting of hormone-sensitive or hormone-status-unknown MBC, endocrine therapy continues to provide favorable outcomes. Once it is determined that endocrine therapy alone is no longer an option in the MBC setting, chemotherapy is re-instituted. Since highly active chemotherapy is the foundation of early invasive breast cancer treatment, limited options remain in the advanced disease setting. Hence, agents that are highly active in pretreated patients and those with low toxicity-to-efficacy ratios are sorely needed. Capecitabine, gemcitabine, nab-paclitaxel, ixabepilone, and eribulin are novel agents that can be utilized in patients with progressive disease. However, regimen, sequence, and dose optimization strategies must be further developed. Finally, the discovery of the tumor-targeting agents, trastuzumab, lapatinib, and bevacizumab, offers hope for women with MBC, but new drugs and combinations that selectively act on specific tumor targets and that improve overall survival with low toxicity are still greatly needed.
Dr Marcus Ravnan is associate professor, department of pharmacy practice, University of the Pacific Thomas J. Long School of Pharmacy and Health Sciences, Stockton, Calif.; Dr Susan Ravnan is visiting lecturer, department of pharmacy practice, University of the Pacific Thomas J. Long School of Pharmacy and Health Sciences; and Dr Walberg is assistant professor, department of pharmacy practice, University of the Pacific Thomas J. Long School of Pharmacy and Health Sciences.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. American Cancer Society. Breast cancer overview guide. Available at http://www.Cancer.org/. Accessed April 4, 2011.
2. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300.
3. Cardoso F, Senkus-Konefka E, Fallowfield L, Costa A, Castigilone M. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(suppl 5):v15–v19.
4. Guarneri V, Conte P. Metastatic breast cancer: therapeutic options according to molecular subtypes and prior adjuvant therapy. Oncologist. 2009;14:645–656.
5. Pagani O, Senkus E, Wood W, et al, on behalf of the ESO–MBC Task Force. International guidelines for management of metastatic breast cancer: can metastatic breast cancer be cured? J Natl Cancer Inst. 2010;102:456–463.
6. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. V.2.2010. Breast Cancer. Accessed April 4, 2011. Available at: http://www.nccn.org/professionals/physician_gls/PDF/breast.pdf.
7. Beslija S, Bonneterre J, Burstein JH, et al, for the Central European Oncology Group (CECOG). Third consensus on medical treatment of metastatic breast cancer. Ann Oncol. 2009;20:1771–1785.
8. Comen E, Fornier M. Algorithms for the treatment of patients with metastatic breast cancer and prior exposure to taxanes and anthracyclines. Clin Breast Cancer. 2010;10(suppl 2):S7–S19.
9. Nahta R, Yu D, Hung M, Hortobagyi G, Esteva F. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280.
10. Ross J, Slodkowska E, Symmans F, Pusztai L, Ravdin P, Hortobagyi G. The HER-2 receptor and breast cancer: ten years of targeted anti–HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368.
11. Burstein H, Prestrud A, Seidenfeld J, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Pract. 2010;6:243-246.
12. Freedman R, Winer E. Adjuvant therapy for postmenopausal women with endocrine-sensitive breast cancer. Breast. 2010;19:69–75.
13. Janni W, Hepp P. Adjuvant aromatase inhibitor therapy: outcomes and safety. Cancer Treat Rev. 2010;36:249–261.
14. Harvey H, Kimura M, Hajba A. Toremifene: an evaluation of its safety profile. Breast. 2006;15:142–157.
15. Sideras K, Ingle J, Ames M, et al. Coprescription of tamoxifen and medications that inhibit CYP2D6. J Clin Oncol. 2010;28:2768-2776.
16. Gibson L, Lawrence D, Dawson C, Bliss J. Aromatase inhibitors for treatment of advanced breast cancer in postmenopausal women. Cochrane Database Syst Rev. 2009;4:003370.
17. Howell A, Robertson J, Abram P, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol. 2004;22:1605–1613.
18. Robertson, J, Come S, Jones S, et al. Endocrine treatment options for advanced breast cancer-the role of fulvestrant. Eur J Cancer. 2005;41:346–356.
19. Howell A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr Relat Cancer. 2006;13:689–706.
20. Valachis A, Mauri D, Polyzos N, Mavroudis D, Georgoulias, Casazza G. Fulvestrant in the treatment of advanced breast cancer: a systematic review and meta-analysis of randomized controlled trials. Crit Rev Oncol Hematol. 2010;73:220–227.
21. Robertson J, Llombart-Cussac A, Rolski J, et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol. 2009;27:4530–4535.
22. Di Leo A, Jerusalem G, Petruzelka L, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2010;28:4594–4600.
23. Jelovac D, Macedo L, Goloubeva O, Handratta V, Brodie A. Additive antitumor effect of aromatase inhibitor letrozole and antiestrogen fulvestrant in a postmenopausal breast cancer model. Cancer Res. 2005;65;5439–5444.
24. Moreno-Aspitia A, Perez E. Treatment options for breast cancer resistant to anthracycline and taxane. Mayo Clin Proc. 2009;84:533–545.
25. Saad E, Katz A, Buyse M. Overall survival and post-progression survival in advanced breast cancer: a review of recent randomized clinical trials. J Clin Oncol. 2010;28:1958–1962.
26. Mauri D, Kamposioras K, Tsali L, et al. Overall survival benefit for weekly vs. three-weekly taxanes regimens in advanced breast cancer: a meta-analysis. Cancer Treat Rev. 2010;36:69–74.
27. Reichardt P, Von Minckwitz G, Thuss-Patience P, et al. Multicenter phase II study of oral capecitabine (Xeloda) in patients with metastatic breast cancer relapsing after treatment with a taxane-containing therapy. Ann Oncol. 2003;14:1227–1233.
28. Fumoleau P, Largillier R, Clippe C, et al. Multicentre, phase II study evaluating capecitabine monotherapy in patients with anthracycline- and taxane-pretreated metastatic breast cancer. Eur J Cancer. 2004;40:536–542.
29. Barrett-Lee P, Bidard F, Pierga J. Contemporary issues and the potential uses of capecitabine in metastatic breast cancer. Cancer Treat Rev. 2009;35:582–589.
30. Kaufmann M, Maass N, Costa S, et al, for the GBG-39 Trialists. First-line therapy with moderate dose capecitabine in metastatic breast cancer is safe and active: results of the MONICA trial. Eur J Cancer. 2010;46:3184-3191.
31. Thomas E, Gomez H, Li R, et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol. 2007;25:5210–5217.
32. Benekli M, Yildiz R, Uner A, et al, for the Anatolian Society of Medical Oncology (ASMO). Gemcitabine plus capecitabine combination in metastatic breast cancer patients previously treated with anthracyclines and taxanes. Oncology. 2007;72: 308-313.
33. Wirk B, Perez E. Role of gemcitabine in breast cancer management: an update. Semin Oncol. 2006;33(1 Suppl 2):S6–S14.
34. Albain K, Nag S, Calderillo-Ruiz G, et al. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26:3950–3957.
35. Blackstein M, Vogel C, Ambinder R, Cowan J, Iglesias J, Melemed A. Gemcitabine as first-line therapy in patients with metastatic breast cancer: a phase II trial. Oncology. 2002;62:2–8.
36. Modi S, Currie V, Seidman A, et al. A phase II trial of gemcitabine in patients with metastatic breast cancer previously treated with an anthracycline and taxane. Clin Breast Cancer. 2005;6:55–60.
37. Gradishar W, Cortes J. Clinical efficacy and emerging therapeutic utilization of novel taxanes. Eur J Cancer. 2008;(suppl 6):12–21.
38. Fernández Y, Cueva J, Palomo A, et al. Novel therapeutic approaches to the treatment of metastatic breast cancer. Cancer Treat Rev. 2010;36:33–42.
39. Henderson I, Bhatia V. Nab-paclitaxel for breast cancer: a new formulation with an improved safety profile and greater efficacy. Expert Rev Anticancer Ther. 2007;7:919–943.
40. Gradishar W, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil–based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803.
41. Gradishar W, Krasnojon D, Cheporov S, et al. Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J Clin Oncol. 2009;27:3611–3619.
42. Gradishar W, Krasnojon D, Cheporov S, et al. Randomized comparison of weekly or every-3-week (q3w) nab-paclitaxel compared to q3w docetaxel as first-line therapy in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol. 2007;25(June 20 suppl)1032.
43. Morris P, Fornier M. Novel anti-tubulin cytotoxic agents for breast cancer. Expert Rev Anticancer Ther. 2009;9:175–185.
44. Steinberg M. Ixabepilone: a novel microtubule inhibitor for the treatment of locally advanced or metastatic breast cancer. Clin Ther. 2008;30:1590–1617.
45. Cortes J. Survival prolongation in metastatic breast cancer. Clin Advances Hematol Oncol. 2010;8(suppl 14):12–15.
46. Roché H, Yelle L, Cognetti F, et al. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, as first-line therapy in patients with metastatic breast cancer previously treated with anthracycline chemotherapy. J Clin Oncol. 2007;25:3415–3420.
47. Denduluri N, Low J, Lee J, et al. Phase II trial of ixabepilone, an epothilone B analog, in patients with metastatic breast cancer previously untreated with taxanes. J Clin Oncol. 2007;25:3421–3427.
48. Perez E, Lerzo G, Pivot X, et al. Efficacy and safety of ixabepilone (BMS-247550) in a phase II study of patients with advanced breast cancer resistant to an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2007;25:3407–3414.
49. Thomas E, Tabernero J, Fornier M, et al. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in patients with taxane-resistant metastatic breast cancer. J Clin Oncol. 2007;25:3399–3406.
50. Thomas E. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment [correspondence]. J Clin Oncol. 2008;26:2223.
51. Orsini L, Mukhopadhyay P, Peck R, Corey-Lisle P. Quality adjusted time without symptoms or toxicities (Q-TWiST) of ixabepilone plus capecitabine versus capecitabine for metastatic breast cancer (MBC) [abstract]. Presented at: the American Society of Clinical Oncology Breast Cancer Symposium; May 29-June 2, 2009; Orlando, FL. Abstract 170.
52. Orsini L, Mukhopadhyay P, Peck R, Corey-Lisle P. Quality adjusted time without symptoms or toxicities (Q-TWiST) of ixabepilone (Ixa) plus capecitabine (cape) versus capecitabine for metastatic breast cancer (MBC) patients with poor prognostic features (PPF). Cancer Res. 2009;69(suppl 3):782s. Abstract 5051.
53. Cigler T, Vahdat L. Eribulin mesylate for the treatment of breast cancer. Expert Opin Pharmacother. 2010;11:1587–1593.
54. Gradishar W. The place for eribulin in the treatment of metastatic breast cancer. Curr Oncol Rep. 2011;13:11–16.
55. Twelves C, Loesch D, Blum J, et al; on behalf of the Study 305 Investigators. A phase III study (EMBRACE) of eribulin mesylate versus treatment of physician's choice in patients with locally recurrent or metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2010;28(June 20 suppl):CRA1004.
56. Pienńkowski T, Zielinski C. Tratuzumab treatment in patients with breast cancer and metastatic CNS disease. Ann Oncol. 2010;21:917–924.
57. Konecny G, Pauletti G, Untch M, et al. Association between HER2, TOP2A, and the response to anthracycline-based chemotherapy in high-risk primary breast cancer. Breast Cancer Res Treat. 2010;120:481–489.
58. Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol. 2009;27:5838–5847.
59. Costanzo F, Gasperoni S, Rotella V, Di Costanzo F. Targeted delivery of albumin bound paclitaxel in the treatment of advanced breast cancer. Onco Targets Ther. 2009;2:179–188.
60. Cobleigh M, Vogel C, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648.
61. Hudis C. Trastuzumab-mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51.
62. Andersson M, Lidbrink E, Bjèrre K, et al. Phase III randomized study comparing docetaxel plus trastuzumab with vinorelbine plus human epidermal growth factor receptor 2–positive breast cancer: the HERNATA study. J Clin Oncol. 2011;29:251-253.
63. Michalaki V, Fotiou S, Gennatas S, Gennatas C. Trastuzumab plus capecitabine and docetaxel as first-line therapy for HER2-positive metastatic breast cancer: phase II results. Anticancer Res. 2010;30:3051–3054.
64. Untch M, Muscholl M, Tjulandin S, et al. First-line trastuzumab plus epirubicin and cyclophosphamide therapy in patients with human epidermal growth factor receptor 2–positive metastatic breast cancer: cardiac safety and efficacy data from the Herceptin, cyclophosphamide, and epirubicin (HERCULES) trial. J Clin Oncol. 2010;28:1473–1480.
65. Extra J, Antoine E, Vincent-Salomon A, et al. Efficacy of trastuzumab in routine clinical practice and after progression for metastatic breast cancer patients: the observational Hermine study. Oncologist. 2010;15:799–809.
66. Scaltriti M, Verma C, Guzman M, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28:803–814.
67. Medina P, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30:1426–1447.
68. Sutherland S, Ashley S, Miles D, et al. Treatment of HER2-positive metastatic breast cancer with lapatinib and capecitabine in the lapatinib expanded access programme, including efficacy in brain metastases-the UK experience. Br J Cancer. 2010;102:995–1002.
69. Burstein H, Storniolo A, Franco S, et al. A phase II study of lapatinib monotherapy in chemotherapy-refractory HER2-positive and HER2-negative advanced or metastatic breast cancer. Ann Oncol. 2008;19:1068–1074.
70. Johnston S, Trudeau M, Kaufman B, et al. Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. J Clin Oncol. 2008;26:1066–1072.
71. Cameron D, Casey M, Oliva C, Newstat B, Imwalle B, Geyer C. Lapatinib plus capecitabine in women with HER-2–positive advanced breast cancer: final survival analysis of a phase III randomized trial. Oncologist. 2010;15:924–934.
72. Geyer C, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743.
73. Schwartzberg L, Franco S, Florance A, O'Rourke L, Maltzman J, Johnston S. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor–positive metastatic breast cancer. Oncologist. 2010;15:122–129.
74. Blackwell K, Burstein H, Storniolo A, et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28:1124–1130.
75. Moy B, Goss P. Lapatinib-associated toxicity and practical management recommendations. Oncologist. 2007;12:756–765.
76. Crown J, Burris H III, Boyle F, et al. Pooled analysis of diarrhea events in patients with cancer treated with lapatinib. Breast Cancer Res Treat. 2008;112:317–325.
77. Weingart S, Brown E, Bach P, et al. NCCN task force report: oral chemotherapy. J Natl Compr Cancer Netw. 2008;6(suppl 3):S1–S14.
78. Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676.
79. Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2009;27:4966–4972.
80. Robert N, Dieras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2–negative locally recurrent or metastatic breast cancer (MBC). J Clin Oncol. 2011 Mar 7, doi:10.1200/JCO.2010.28.0982 [Epub ahead of print].
81. Miller K, Chap L, Holmes F, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–799.
82. Miles D, Chan A, Dirix L, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor–2 negative metastatic breast cancer. J Clin Oncol. 2010;28:3239–3247.
83. Smith I, Pierga J, Biganzoli L, et al, on behalf of the ATHENA Study Group. First-line bevacizumab plus taxane-based chemotherapy for locally recurrent or metastatic breast cancer: safety and efficacy in an open-label study in 2251 patients. Ann Oncol. 2011;22:595-602.
84. Valachis A, Polyzos N, Patsopoulos N, Georgoulis V, Mavroudis D, Mauri D. Bevacizumab in metastatic breast cancer: a meta-analysis of randomized controlled trials. Breast Cancer Res Treat. 2010;122:1–7.
85. FDA news teleconference, Dec. 16, 2010. Avastin (bevacizumab): process for removal of breast cancer indication begun. Accessed April 4, 2011. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm237280.htm.







