
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Milnacipran: A serotonin and norepinephrine reuptake inhibitor for the management of fibromyalgia syndrome
Milnacipran was recently approved by FDA for the management of fibromyalgia syndrome. Clinical trials have demonstrated that this agent may be a reasonable alternative or adjunct therapy for patients with fibromyalgia syndrome who are unable to tolerate first-line tricyclic antidepressant therapy.



Key Points
Abstract
Fibromyalgia syndrome (FMS) is a condition classified by widespread chronic pain and is associated with fatigue, morning stiffness, and disordered sleep. Milnacipran is a serotonin and norepinephrine reuptake inhibitor (SNRI) that was recently approved by FDA for the management of FMS. In clinical trials, milnacipran has demonstrated efficacy in reducing FMS symptoms, with patients exhibiting response rates of 25% to 33%. Milnacipran is generally well tolerated and has minimal interactions with concomitant drug therapies. Nausea was the adverse event most frequently reported in clinical trials. In patients with FMS who are unable to tolerate first-line tricyclic antidepressant therapy, milnacipran may be a reasonable alternative or adjunct therapy. (Formulary. 2009;44:197–202.)
Fibromyalgia syndrome (FMS) is a rheumatologic disorder characterized by chronic nonlocalized pain and tenderness. The chronic pain condition is classified by the American College of Rheumatology (ACR) as ≥3 months of widespread pain in combination with tenderness at ≥11 of 18 defined tender point sites.1 In addition to chronic pain, 73% to 85% of patients with FMS also report experiencing fatigue, morning stiffness, and sleep disorders.1 Patients may also experience myriad auxiliary symptoms, such as irritable bowel or bladder, depression, anxiety, paresthesias, headaches, and cognitive disturbances.2
Antidepressant agents offer unique mechanisms within the neurohormonal system that may alleviate symptoms that commonly affect quality of life in patients with FMS. Tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin and norepinephrine reuptake inhibitors (SNRIs) have been associated with reductions in pain, fatigue, and disordered sleep independent of reduction in depression in patients with FMS.4,5 TCAs have been considered first-line therapy in treating FMS, but their use is limited by their anticholinergic side-effect profile.
Milnacipran (Savella, Forest/Cypress) is an orally administered SNRI that was recently approved for the management of FMS.5,6
CLINICAL PHARMACOLOGY
Milnacipran ([±]-[1R(S),2S(R)]-2-[aminomethyl]-N, N-diethyl-1-phenyl-cyclopropanecarboxamide hydrochloride) is an SNRI that has demonstrated greater inhibition of norepinephrine than of serotonin in rat hypothalamus.6 This agent also has some affinity for the N-methyl-D-aspartate (NMDA) receptor but does not bind to dopamine, histamine, or muscarinic receptors and thus lacks many of the side effects of TCAs.6,7 Magnetic resonance imaging (MRI) scans performed after administration of milnacipran have demonstrated increased brain activity in specific areas of the brain (caudatus nucleus, anterior insula, anterior cingulum, and amygdala) that are involved in the pain response in FMS.8
PHARMACOKINETICS

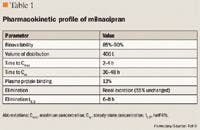
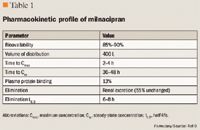
CLINICAL TRIALS

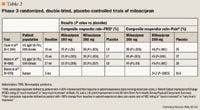
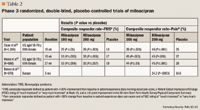
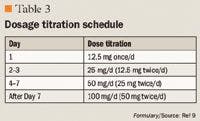
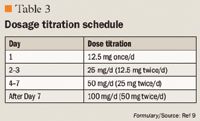
Clauw et al10 conducted a study of 1,196 patients with FMS across 86 US centers. This was a registration trial to evaluate the efficacy and safety of milnacipran in a larger patient population. Eligible patients were required to have a score ≥4 on the Fibromyalgia Impact Questionnaire (FIQ) and a VAS pain score of ≥40 (scale, 0–100). All patients had an FMS diagnosis based on ACR criteria and had to discontinue all CNS-active therapy, as well as medications that would interfere with the evaluation of FMS symptoms; however, patients were allowed to use hydrocodone as rescue therapy (maximum dose, 60 mg/d). Patients entered a 1- to 4-week washout period, followed by a 2-week baseline assessment period. Eligible patients were then randomized to treatment with milnacipran 100 mg/d, milnacipran 200 mg/d, or placebo. Milnacipran-treated patients received 12.5 mg on Day 1, 12.5 mg twice/d for 2 days, 25 mg twice/d for 4 days, and 50 mg twice/d for 7 days. Patients who were randomized to the 200-mg/d group received 100 mg twice/d for 7 days. All other randomized patients underwent placebo dose-escalation to maintain blinding. All patients remained on a stable dose from Weeks 3 to 15, and any patient who was intolerant of the assigned dose was dropped from the trial.
At study end, significantly more patients treated with milnacipran than those treated with placebo met the 3 criteria for improvement (changes in morning-recall VAS pain scores, global status on PGIC, and SF-36 PCS score) in FMS symptom response (observed cases [OC] analysis: milnacipran 100 mg/d, 25%; P≤.01 vs placebo; milnacipran 200 mg/d, 26%; P≤.001 vs placebo; placebo, 13%).10 Both doses of milnacipran significantly decreased the composite pain response versus placebo (OC analysis: milnacipran 100 mg/d, 39%; milnacipran 200 mg/d, 46%; placebo, 25%; P≤.001 for both vs placebo). There were also significant differences with both doses of milnacipran in secondary outcomes, including time-weighted averages of weekly mean PED morning-recall pain scores (last observation carried forward [LOCF] analysis, mean ± standard error of the mean [SEM]: milnacipran 100 mg/d, 44.3±0.9; milnacipran 200 mg/d, 43.3±1.0; placebo, 48.9±0.9; P≤.001 for both vs placebo) and PGIC scores (LOCF analysis, mean ± SEM: milnacipran 100 mg/d, 3.1±0.1; milnacipran 200 mg/d, 3.0±0.1; placebo, 3.5±0.1; P≤.001 for both vs placebo). Time-weighted averages of SF-36 PCS reached statistical significance only in the milnacipran 100-mg/d group (LOCF analysis, mean ± SEM: milnacipran 100 mg/d, 35.3±0.4; P≤.001 vs placebo).
The most common adverse effect was nausea, which occurred in 34.3%, 37.6%, and 19.2% of patients treated with milnacipran 100 mg/d, milnacipran 200 mg/d, and placebo, respectively.10 Other adverse effects that occurred more frequently with milnacipran than with placebo were constipation, hot flushing, dizziness, palpitations, hyperhidrosis, hypertension, vomiting, tachycardia, and migraine. More patients treated with milnacipran 100 or 200 mg/d discontinued therapy because of adverse effects than those treated with placebo (19.5%, 23.7%, and 9.5%, respectively). The 2 milnacipran doses demonstrated similar efficacy; however, more adverse events were reported in patients receiving milnacipran 200 mg/d. This study demonstrated that milnacipran not only had an effect on pain but also improved fatigue and patient-reported cognitive complaints.
Mease et al11 randomized 888 patients from 59 centers in the United States to treatment with milnacipran 100 mg/d, milnacipran 200 mg/d, or placebo for 6 months. Patients had to discontinue all CNS active therapies but could continue taking acetaminophen, aspirin, and stable doses of nonsteroidal anti-inflammatory drugs (NSAIDs). Patients could use hydrocodone ≤60 mg/d as rescue therapy; however, they could not use this therapy for 48 hours before study visits or during each 2-week period preceding the primary end point (Weeks 14–15 and 26–27). Patients were excluded from the study if they had a severe psychiatric illness. Eligible patients entered a 1- to 4-week washout period and then entered a 2-week baseline period. A greater proportion of patients was randomized to milnacipran 200 mg/d than to milnacipran 100 mg/d or placebo for the collection of long-term safety data. All patients underwent a dose-escalation period of 3 weeks, including those treated with placebo to ensure blinding. Milnacipran-treated patients were scheduled to receive 24 weeks of a stable dose of milnacipran (27-week exposure in total).
At 15 weeks, a greater proportion of patients treated with milnacipran 100 and 200 mg/d had met the criteria for a composite responder than those treated with placebo (OC analysis: milnacipran 100 mg/d, 32.8%; P≤.003 vs placebo; milnacipran 200 mg/d, 32.8%; P≤.001 vs placebo; placebo, 17.3%). At 6 months, 31.9% (P≤.017 vs placebo) of patients treated with milnacipran 200 mg/d and 33.3% (P≤.056) of those treated with milnacipran 100 mg/d were composite FMS responders compared with 19% of placebo-treated patients (OC analysis). A statistically significant reduction in pain was observed within 1 week of initiation of milnacipran therapy in both treatment groups and continued throughout the duration of the study, with maximal pain relief observed at 9 weeks. In the OC analysis, 45.2% (P=.003 vs placebo) of patients treated with milnacipran 100 mg/d and 45.4% (P<.001 vs placebo) of patients treated with milnacipran 200 mg/d were FMS pain composite responders versus 27.2% of patients treated with placebo. Milnacipran-treated patients also experienced improvements in physical functioning, fatigue, and cognition as measured by the SF-36 domains of physical functioning, Multidimensional Fatigue Inventory (MFI) total score, and Multiple Ability Self-report Questionnaire (MASQ) score. There were statistically significant improvements in physical functioning (mean change from baseline, 3.55; P<.05 vs placebo), bodily pain (mean change from baseline, 6.05; P<.01 vs placebo), and mental health (mean change from baseline, 3.70; P<.01 vs placebo) as measured by the SF-36 in the milnacipran 200-mg/d group versus placebo. Milnacipran 200 mg/d was also associated with a statistically significant mean change from baseline in fatigue versus placebo at 15 and 27 weeks (MFI score: milnacipran 200 mg/d, –5.62 at Week 15 and –5.80 at Week 27; placebo, –3.04 at Week 15 and –3.35 at Week 27; P<.05). Mean changes in patients' baseline perception of cognitive function were greater in the milnacipran 200-mg/d group versus the placebo group (milnacipran 200 mg/d, –2.28 at Week 15 and –2.68 at Week 27; placebo, 0.10 at Week 15 and 0.16 at Week 27; P<.05). There was no difference among groups with respect to sleep quality or quantity as measured by the Medical Outcomes Study (MOS)-Sleep Problems Indices.
At 6 months, 43%, 46%, and 35% of patients in the milnacipran 100-mg/d, milnacipran 200-mg/d, and placebo groups, respectively, had discontinued therapy. Most patients discontinued therapy because of adverse events (milnacipran 100 mg/d, 19.6%; milnacipran 200 mg/d, 27%; placebo, 10.3%).11 The overall incidence of side effects was 90.7%, 83.9%, and 85.2% in patients treated with milnacipran 200 mg/d, milnacipran 100 mg/d, and placebo, respectively. The most commonly reported adverse event was nausea, consistent with other clinical trials. This trial and the trial by Clauw et al10 may have limited applicability to patients with FMS who have psychiatric disorders, as these patients were excluded from these studies. It is not known how many patients were excluded because of psychiatric illness in each study; however, in the study by Clauw et al,10 53% of screened patients were excluded, and in the study by Mease et al,11 46% of screened patients were excluded.
Branco et al12 conducted a phase 3 European trial involving 678 patients at 83 centers. Eligible patients were required to have a mean daily VAS pain score of 20 to 90, FIQ-Physical Function (PF) subscore ≥3, and a Beck Depression Inventory (BDI) score ≤25 at baseline. Patients were randomized to milnacipran 100 mg twice/d or placebo for 12 weeks after a 4-week dose escalation. Patients underwent a 9-day down-titration after 12 weeks of therapy and were assessed at 2 weeks after treatment. The primary end point was pain composite responder rate and the PGIC at 16 weeks. The primary end point was measured in the same way as in Clauw et al10 and Mease et al.11 Patients were excluded if they had a severe psychiatric illness; eligible patients had to discontinue all CNS active therapies, as in previous trials.10,11 A major secondary end point was FIQ total score as measured by weekly recall self-assessment at on-site visits.
Milnacipran was associated with a higher composite response rate than was placebo (24.2% vs 14.6%; OR=1.9; P=.0003). The FIQ-Total score was significantly improved in patients treated with milnacipran versus those treated with placebo (adjusted mean difference, –3; P=.015). Patients treated with milnacipran demonstrated improvements in pain (morning VAS, P<.0001 vs placebo), quality of life (both mental and physical, as measured by FIQ-Total-Modified and SF-36 mental component score [MCS], P<.01 vs placebo), fatigue (MFI-Total, P<.01 vs placebo), global status (PGIC, P<.0001 vs placebo), and cognition (MASQ-Total, P<.05 vs placebo). Withdrawal of treatment because of adverse events was 22.3% and 9.9% in the milnacipran and placebo groups, respectively. This study demonstrated that milnacipran 200 mg/d improved pain and overall functioning for patients with FMS with minimal side effects.
ADVERSE EVENTS
Milnacipran was well tolerated in clinical trials. The most frequently reported adverse event was nausea, occurring in 32% to 40% of patients.13 Other common adverse events (incidence ≥5%) were noradrenergic in nature, such as dry mouth, hyperhidrosis, headache, hot flush, and constipation.7,9 In clinical trials, the incidence of early discontinuation of milnacipran because of adverse events was 22% to 27%.10–12
Compared with TCAs, milnacipran may have fewer anticholinergic and alpha-adrenergic effects.7 Milnacipran may have more noradrenergic effects but fewer serotonergic effects than duloxetine or venlafaxine.7 Milnacipran is unlikely to have a significant effect on cardiac function. In studies, palpitations were reported in 7% to 8% of milnacipran-treated patients.9 This agent does not significantly increase blood pressure, unlike venlafaxine, and has only a minimal association with increases in heart rate.7,9 However, increases in blood pressure and heart rate may occur, and manufacturer labeling recommends monitoring of these parameters.9



DRUG INTERACTIONS
Milnacipran is unlikely to have many pharmacokinetic interactions with concomitant drug therapies because this agent undergoes minimal CYP450 metabolism, is mostly excreted unchanged in the urine, and has low plasma protein binding. Milnacipran may, however, interact with other drugs from a pharmacodynamic perspective.9 Serotonin syndrome may occur when milnacipran is administered with lithium, which impairs serotonin metabolism, or when milnacipran is administered with serotonergic drugs.9 Because milnacipran inhibits reuptake of norepinephrine, caution should be exercised when milnacipran is administered with epinephrine and norepinephrine, as concomitant use may lead to paroxysmal hypertension and arrhythmia; milnacipran may also oppose clonidine's antihypertensive effect.9 Concomitant milnacipran and intravenous digoxin use should be avoided, as coadministration of these agents has been reported to cause postural hypotension and tachycardia.9 Reports of postural hypotension and euphoria have also been documented when clomipramine is replaced with milnacipran.9 As milnacipran is centrally acting, general caution should be exercised when this agent is used with other centrally acting drugs. Concomitant use with monoamine oxidase inhibitors (MAOIs) is contraindicated.9
DOSING AND ADMINISTRATION

Dr Cios and Dr Kim are senior clinical pharmacists, Brigham and Women's Hospital, Boston, Massachusetts.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
In each issue, the "Focus on" feature reviews a newly approved or investigational drug of interest to pharmacy and therapeutics committee members. The column is coordinated by Robert A. Quercia, MS, RPh, clinical manager and director of Drug Information, Department of Pharmacy Services, Hartford Hospital, Hartford, Conn, and adjunct associate professor, University of Connecticut School of Pharmacy, Storrs, Conn; and by Craig I. Coleman, PharmD, assistant professor of pharmacy practice, University of Connecticut School of Pharmacy, and director, Pharmacoeconomics and Outcomes Studies Group, Hartford Hospital.
EDITORS' NOTE: The clinical information provided in "Focus on" articles is as current as possible. Due to regularly emerging data on developmental or newly approved drug therapies, articles include information published or presented and available to the author up until the time of the manuscript submission.
REFERENCES
1. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia: Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33:160–172.
2. Lawson K. Emerging pharmacological therapies for fibromyalgia. Curr Opin Investig Drugs. 2006; 7:631–636.
3. Wolfe F, Ross K, Anderson J, Russell IJ, Hebert L. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38:19–28.
4. Crofford LJ. Pain management in fibromyalgia. Curr Opin Rheumatol. 2008;20:246–250.
5. Uceyler N, Hauser W, Sommer C. A systematic review on the effectiveness of treatment with antidepressants in fibromyalgia syndrome. Arthritis Rheum. 2008;59:1279–1298.
6. Owen RT. Milnacipran hydrochloride: Its efficacy, safety and tolerability profile in fibromyalgia syndrome. Drugs Today (Barc). 2008;44:653–660.
7. Leo RJ, Brooks VL. Clinical potential of milnacipran, a serotonin and norepinephrine reuptake inhibitor, in pain. Curr Opin Investig Drugs. 2006;7:637–642.
8. Gracely RH, Jensen K, Petzke F, et al. The effect of milnacipran on pain modulatory systems in fibromyalgia: An fMRI analysis [abstract]. Ann Rheum Dis. 2008;67(suppl 2):255.
9. Savella [package insert]. New York, NY: Forest Pharmaceuticals, Inc.; 2009.
10. Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: A 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial [erratum in Clin Ther. 2009;31:446]. Clin Ther. 2008;30:1988–2004.
11. Mease PJ, Clauw DJ, Gendreau RM, et al. The efficacy and safety of milnacipran for treatment of fibromyalgia. A randomized, double-blind, placebo-controlled trial [erratum in J Rheumatol. 2009;36:661]. J Rheumatol. 2009;36:398–409.
12. Branco JC, Perrot S, Bragee G, et al. Milnacipran for the treatment of fibromyalgia syndrome: A European multicenter, randomized, double-blind, placebo-controlled trial [abstract]. Ann Rheum Dis. 2008;67(suppl 2):251.
13. Vitton O, Gendreau M, Gendreau J, Kranzler R, Rao SG. A double-blind placebo-controlled trial of milnacipran in the treatment of fibromyalgia. Hum Psychopharmacol. 2004;19(suppl 1):S27–S35.
14. Gendreau RM, Thorn MD, Gendreau JF, et al. Efficacy of milnacipran in patients with fibromyalgia. J Rheumatol. 2005;32:1975–1985.

Employers Face Barriers With Adopting Biosimilars
March 1st 2022Despite the promise of savings billions of dollars in the United States, adoption of biosimilars has been slow. A roundtable discussion among employers highlighted some of the barriers, including formulary design and drug pricing and rebates.






