
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Oral oncolytics: assessing value of newer agents versus current standards of care as part of P&T processes
Oral oncolytics are relatively new to the field of cancer therapy. However, they now make up about 25% of the oncology market and their use is continually expanding. The current insurance system is not efficiently equipped to handle their rapid entry into the market.
ABSTRACT
Oral oncolytics are relatively new to the field of cancer therapy. They currently make up about 25% of the oncology market and their use is continually expanding. The current insurance system is not efficiently equipped to handle the rapid expansion of oral oncolytics into the market, and the current insurance benefit design contributes significantly to access issues for patients. This article offers formulary decision makers information needed to evaluate newer oral oncolytic therapies compared with existing standards of care using guidelines from the National Comprehensive Cancer Network®. The oral agents discussed are limited to those introduced into the market since 2007.(Formulary. 2013; 48:258-265)

Click to view infographic
Oral oncolytics are relatively new to the field of cancer therapy. However, they now make up about 25% of the oncology market and their use is continually expanding.1,2 The current insurance system is not efficiently equipped to handle their rapid entry into the market.1 Due to the increase in multi-tier formularies, the growth in outpatient medication spending decreased in recent years while the demand for specialty drugs, including oral oncolytics, has continued to accelerate.3 Pharmacies may not stock oral oncolytic agents due to high cost, and some physicians have more incentive to prescribe intravenous (IV) medications under the current reimbursement system.4 The current insurance benefit design contributes significantly to the access issues patients encounter when attempting to obtain oral oncolytic agents.3 As insurers consider a variety of payment and distribution strategies to regulate the use and cost of oral oncolytics, more patients are being placed in financial turmoil to pay for their expensive medications, thus putting patients at risk for noncompliance.3 Providers, pharmacists, and patients are also facing the administrative burden of dealing with patient assistance programs and insurance plans.5
Medicare and other insurers have distinct medical and pharmacy benefits.6 The medical benefit in Medicare ensures that physician services, including physician-administered drugs, and hospital services are covered.6 Meanwhile, the pharmacy benefit usually covers self-administered drugs including oral medications and some subcutaneous injectables.6 This bifurcated insurance setting can create artificial enticements for physicians to prescribe IV medications and hinder the use of oral oncolytics.6 In addition, differences in cost-sharing among patients can cause financial difficulties by making the prescription drug benefit inaccessible for those that only have medical benefit coverage.6 One study found that 1 in 4 patients who filled their prescriptions and incurred over $500 in out-of-pocket expenses did not return to pick it up or follow up with a new oncology medication within 90 days.4
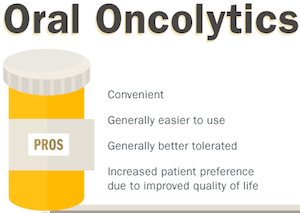
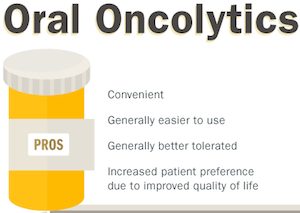
This conundrum has led to insurers trying to re-evaluate how they can pay for oral oncolytics through the incorporation of clinical and evidence-based guidelines. They realize that there are certain advantages to increasing the incentive for providers to utilize oral oncolytics while making them cost effective and accessible to patients. For example, oral oncolytics are generally easy to administer and do not require office visits, thus making them more convenient for patients. In comparison to IV formulations, oral oncolytics are also generally better tolerated,7 and patients are increasingly showing a preference for oral chemotherapy due to an improved quality of life.7
This article offers formulary decision makers information needed to evaluate newer oral oncolytic therapies compared with standard therapy using the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Comparisons of newer oral oncolytics versus injectables in patients with breast, lung, prostate, and colorectal cancer will be discussed in Part 2 of this article. The oral agents discussed are limited to those introduced into the market since 2007 (Table 1).
NEWER ORAL ONCOLYTIC AGENTS COMPARED WITH STANDARD THERAPY
The newly approved oral oncolytic therapies from Table 1 will be compared to existing or older oral oncolytics classified as category 1 by the NCCN Guidelines® for Chronic Myelogenous Leukemia (CML), Kidney Cancer, Medullary Thyroid Carcinoma (MTC), and Melanoma.
Chronic myelogenous leukemia
CML is characterized by a reciprocal translocation between chromosomes 9 and 22 resulting in the formation of the Philadelphia (Ph) chromosome, which is manifested in patients with CML.8,9 This translocation t(9;22) results in the head-to-tail fusion of the breakpoint cluster region (BCR) gene on chromosome 22 at band q11 and the Abelson murine leukemia (ABL) gene located on chromosome 9 at band q34. The product of the BCR-ABL fusion gene (p210, a fusion protein with deregulated tyrosine kinase activity) is believed to play a central role in the initial development of CML.8,9 Therefore, imatinib, dasatinib, nilotinib, and bosutinib play an important role in the management of CML through the inhibition of BCR-ABL tyrosine kinase. The measurement of hematologic, cytogenetic, and molecular responses controls how a patient will respond to tyrosine kinase inhibitors (TKIs).8,9 Achieving a complete cytogenetic response (CCyR) within a year after initiation of therapy and eventually a major molecular response (MMR) while preventing disease progression to accelerated or blast phase is the main goal of therapy.8,9 Complete hematologic response (CHR), as defined by Faderl et al, includes the complete normalization of peripheral blood counts, with leukocyte count <10´109/L, a platelet count <450´109/L, and no immature cells such as myelocytes, promyelocytes, or blasts in the peripheral blood.8,9 O’Brien and colleagues noted that a CCyR indicates there are no Ph-positive metaphases.8,10 Hughes et al defined the complete molecular response (CMR) as no detectable BCR-ABL mRNA by real-time quantitative polymerase chain reaction using International Scale.8,11
Imatinib mesylate inhibits the BCR-ABL tyrosine kinase.12 The IRIS trial demonstrated an overall survival (OS) of 89% and a freedom from progression to accelerated or blast phase of 91% at 4 years after initial treatment with imatinib.13 The CHR, major cytogenetic response (MCyR), and CCyR were 93%, 86% and 81%, respectively, after a follow-up of 4.5 years on imatinib.13 Adverse events (AEs) reported in 40% or more participants were fluid retention, nausea, musculoskeletal pain, and rash, while hematologic AEs were neutropenia, thrombocytopenia, and anemia.13
Dasatinib is an inhibitor of ABL and SRC family of kinases with an ability to bind to both the active and inactive conformation of the ABL kinase domain.14 This provides an advantage by making it active to mutations resistant to imatinib.14 Shah et al demonstrated that 100 mg dasatanib once daily was equally as effective as 70 mg twice daily.15 After 2 years, patients on dasatinib had achieved a CCyR (50% vs. 54%), MCyR (63% vs. 61%), progression-free survival (PFS) (80% vs. 76%), and OS (91% vs. 88%).15 Fewer patients on 100 mg once daily had grade 3 to 4 AEs such as pleural effusions and thrombocytopenia.15 They were also less likely to discontinue from the study due to toxicity or to require dose reductions and interruptions.15
Nilotinib is 20 to 50 times more potent in imatinib-resistant cell lines and is a highly selective inhibitor of BCR-ABL tyrosine kinase.8 Nilotinib 300 mg and 400 mg twice daily was compared to imatinib 400 mg once daily in a long-term follow-up trial of the Evaluating Nilotinib Efficacy and Safety in Clinical Trials newly diagnosed patients (ENESTnd) study.16 The MMR of nilotinib (73% and 70%) was significantly higher than imatinib (53%, P<0.0001) with an estimated 3-year PFS rate of 99.3%, 98.7%, and 95.2% for all 3 treatment groups.16 Twenty-nine percent of patients exhibited grade 3 to 4 thrombocytopenia and neutropenia.16 QT prolongation was noted in patients on nilotinib, and the recommendation was to avoid QT-prolonging drugs and to correct electrolytes before beginning therapy.16
Bosutinib is a BCR-ABL SRC inhibitor with little activity against stem cell factor receptor (c-KIT) and platelet-derived growth factor receptors (PDGFR-α and –β).17 Khoury et al noted estimated PFS and OS rates at 2 years of 73% and 83% respectively, while CHR, MCyR, and CCyR was seen in 73%, 32%, and 24% after a median follow-up of 28.5 months.17 The most common grade 3 to 4 hematologic AEs were thrombocytopenia, neutropenia, and anemia; diarrhea, nausea, vomiting, and rash were the most common nonhematologic AEs.17
Formulary considerations
CHR, MCyR, and CCyR markers of efficacy are important in evaluating the addition of bosutinib to the formulary. Bosutinib is given 500 mg orally once daily with food. Bosutinib is a good addition to the formulary because, in comparison with other TKIs, it has been associated with minimal cardiac effects such as pericardial effusions and pericarditis, minimal musculoskeletal events, and low incidence of pleural effusions and QT prolongation.8 Nevertheless, dose adjustments are required in patients with grade 3 to 4 diarrhea, liver transaminases greater than 5 times the institutional upper limit of normal (ULN), grade 3 to 4 neutropenia (absolute neutrophil count <1,000/mm3), and grade 3 to 4 thrombocytopenia (platelet count <50,000/mm3). Bosutinib is a more potent BCR-ABL inhibitor and is equally efficacious to dasatinib and nilotinib in patients resistant to or intolerant of imatinib.18 It is also indicated for those patients who have dasatinib mutation F317L and nilotinib mutations Y253H and F359.17 Bosutinib had a higher estimated PFS and OS than imatinib, nilotinib, and dasatinib and it is also better tolerated. However, the phase 3 Bosutinib Efficacy and Safety in Newly Diagnosed Chronic Myeloid Leukemia (BELA) trial did not achieve its primary endpoint of CCyR at 12 months when comparing bosutinib 500 mg once daily to imatinib 400 mg once daily.19 Therefore, it is not recommended as a first line in newly diagnosed patients. However, it can be recommended as second line for those who have failed prior TKI therapies.19
Metastatic renal cell carcinoma (mRCC)
Patients will usually present with a suspicious mass involving the kidney that was diagnosed using either an abdominal/pelvic computerized tomographic (CT) scan or an ultrasound.20 The mainstay of therapy for localized disease (stages I, II, and III) is a radical nephrectomy or nephron-sparing nephrectomy.20 Primary treatment of non-surgically resectable advanced disease with cytokine therapy such as interleukin 2 or interferon provides modest benefit with significant toxicity. Therefore, targeted therapy has become useful as first- and second-line treatments for patients with predominant clear cell histology.20 Assessment of survival is based on the Memorial Sloan-Kettering Cancer Center prognostic factor model. They include 5 variables for risk: interval from diagnosis to treatment of <1 year, Karnofsky performance status <80%, serum hemoglobin less than the lower limit of normal, corrected calcium greater than the ULN, and serum lactate dehydrogenase >1.5´ ULN. Patients with no risk factors are considered low risk while those with 3 or more risk factors are poor risk; intermediate risk is categorized by 1 or 2 risk factors.20
Sunitinib is a multikinase inhibitor targeting several tyrosine kinases including vascular endothelial growth factor receptors (VEGFR-1, -2, and -3), PDGFR-α and -β, c-KIT, FMS-like tyrosine kinase (FLT-3), colony-stimulating factor (CSF-1R), and rearranged during transfection kinase (RET).21 Ninety percent of the patients in this trial had low or intermediate risk and had also undergone nephrectomy before participating. Those on sunitinib fared better, with a median PFS of 11 months compared to 5 months for the interferon alpha arm.22 Sunitinib patients also experienced an OS advantage and higher objective response rate (ORR) over interferon alfa (26.4 vs. 21.8 months, hazard ratio [HR] 0.821, 95% CI 0.673 to 1.001, P=0.051; and 47% vs. 12%, P<0.001, respectively).22 Patients on interferon alfa had grade 3 to 4 toxicity of fatigue, while those on sunitinib reported grade 3 to 4 AEs of neutropenia, thrombocytopenia, diarrhea, hand-foot syndrome (HFS), and hypertension.22
Pazopanib is an oral angiogenesis inhibitor targeting VEGFR-1, -2, -3, PDFGR-α and -β, and c-KIT.23 PFS was significantly prolonged with pazopanib compared with placebo in the overall study population (median PFS, 9.2 vs. 4.2 months; HR, 0.46; 95% CI, 0.34 to 0.62; P<0.0001), in the treatment group with no prior therapy (median PFS, 11.1 vs. 2.8 months; HR, 0.40; 95% CI, 0.27 to 0.60; P<0.0001), and in the treatment group with prior cytokine therapy (median PFS, 7.4 vs. 4.2 months; HR, 0.54; 95% CI, 0.35 to 0.84; P<0.001).23 The ORR was 30% with pazopanib compared with 3% with placebo (P<0.001).23 Sternberg and colleagues reported diarrhea, hypertension, hair color changes, nausea, anorexia, vomiting, fatigue, weakness, abdominal pain, headache, and hepatotoxicity in at least 10% of their patients.23
Axitinib is a selective second-generation inhibitor of VEGFR-1, -2, and -3.24 Overall median PFS was 6.7 months for axitinib 5 mg orally twice daily versus 4.7 months for sorafenib 400 mg twice daily (HR, 0.665; 95% CI, 0.544 to 0.812; P<0.0001).25 The PFS favored axitinib in both groups pretreated with interferon alfa (12.1 vs. 6.5 months; P<0.0001) and with sunitinib (4.8 vs. 3.4 months; P=0.01).25 Hypertension and fatigue were more commonly associated with axitinib, while HFS and alopecia were commonly associated with sorafenib, and diarrhea common to both.25
Formulary considerations
The sixth targeted therapy and third TKI that has received FDA approval for the treatment of mRCC is pazopanib.26 Its AE profile is similar to other TKIs used for the treatment of mRCC, but differences in AE rates and extent may exist.26 It is administered on an empty stomach at a dose of 800 mg daily until disease progression, but dose reduction may be required in patients with baseline elevation of hepatic function tests, particularly total bilirubin.26 A patient with baseline hepatic dysfunction should receive a maximum dose of 200 mg daily depending on the severity. It is primarily metabolized by cytochrome P450 (CYP)3A4, so it should be cautiously administered with inducers and inhibitors of these isoenzymes.26 A noninferiority trial, COMPARZ, of sunitinib versus pazopanib reported that pazopanib was better tolerated than sunitinib; however, the two drugs had similar efficacy.27 Therefore, pazopanib among other therapies including sunitinib, temsirolimus, and bevacizumab plus interferon-α may be considered a first-line treatment option.20 Pazopanib can be given after a patient has failed cytokine therapy but not after another targeted therapy due to limited available data.26 The patient’s individual comorbidities and preferences and varying incidences of AEs should be taken into account when choosing between the TKIs.26
Axitinib is administered as 5 mg twice daily orally with food.28 It is metabolized primarily in the liver via CYP3A4/5.28 Co-administration with CYP3A4 and 1A2 inducers is contraindicated, and those receiving a strong CYP3A4/5 inhibitor or who have hepatic impairment should receive half the dose.28 Additionally, proton pump inhibitors have been shown to reduce the rate of axitinib absorption. Two randomized phase 3 clinical trials reported a significant PFS in axitinib patients who had previously received sunitinib or cytokine therapy, making it a good alternative as a second-line therapy.28 Nevertheless, axitinib is cheaper than sorafenib and significantly cheaper than sunitinib with a survival benefit over both.
Medullary thyroid carcinoma
MTC is a malignancy of the parafollicular C cells of the thyroid, which presents in a sporadic or hereditary pattern with associated RET proto-oncogene mutations.29 A new class of TKIs has been developed to target these RET mutations in selected patients with advanced MTC.29 Patients with advanced MTC have few treatment options; radioactive iodine is not recommended, and chemotherapy is not very effective. Vandetanib and cabozantinib are TKIs that have been shown to increase PFS in patients with this aggressive disease. Surgery is the main treatment for MTC because there is no known systemic cure for unresectable disease.29 The overall goals of treatment vary depending on the type of patient. In asymptomatic patients with RET mutations but no evidence of MTC, the goal is to prevent the onset of disease with prophylactic total thyroidectomy (at the appropriate time) and thus increase survival. However, in patients who present with MTC, the goal is to increase survival with immediate thyroidectomy and to decrease residual disease. In patients with persistent or recurrent disease, the goal is to treat symptomatic locoregional or metastatic disease and to provide palliation.29 Vandetanib and cabozantinib are multikinase inhibitors that have shown activity against RET and other tyrosine kinases in the treatment of unresectable MTC.29 These agents have been shown to decrease tumor burden in select patients with advanced disease.30–32
Vandetanib is an oral receptor multikinase inhibitor targeting epidermal growth factor receptor (EGFR), VEGF, and RET.30 Projected median PFS for vandetanib was 30.5 months, versus actual median PFS of 19.3 months for placebo (HR, 0.46; 95% CI, 0.31 to 0.69; P<0.001).30 Objective response rate (P <0.001), disease control rate (P = 0.001), and biochemical response (P <0.001) were also statistically significant. Overall survival data were immature at data cutoff (HR, 0.89; 95% CI, 0.48 to 1.65). Final analysis of OS will be done when 50% of the patients are dead.30 Most common AEs reported in greater than 25% of the patients on vandetanib included diarrhea, rash, nausea, hypertension, and headache 30
Cabozantinib is an oral receptor TKI targeting tyrosine kinase activity of MET, VEGFR-2, and RET.31 A statistically significant prolongation in PFS was demonstrated among patients treated with cabozantinib compared to those receiving placebo (HR, 0.28; 95% CI, 0.19 to 0.40; P<0.0001), with median PFS times of 11.2 months and 4.0 months and (ORR of 28% vs. 0%, P<0.0001) in the cabozantinib and placebo arms, respectively.31-2 There is a median duration of response for patients in the cabozantinib group with no significant improvement in the OS.31-2 Significant AEs reported included diarrhea, HFS, fatigue, hypocalcemia, and hypertension.31-2
Formulary considerations
Vandetanib is given 300 mg once daily with dosage adjustments required for impaired renal function (CrCl <50), QTcF prolongation (>500 ms), and other grade 3 to 4 toxicities including diarrhea, hypertension, and skin reactions associated with the drug. Vandetanib should not be used in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome.30 These electrolytes should be corrected and periodically monitored while the patient is receiving vandetanib.30 ECGs should be obtained at baseline, 2 to 4 weeks, and 8 to 12 weeks after starting treatment and every 3 months thereafter.30 Its long half-life of 19 days makes it difficult to resolve the prolonged QTc interval.30 Vandetanib does, however, have a more serious AE of cardiotoxicity associated with QT prolongation in comparison to cabozantinib. Due to this, vandetanib is only available through a Risk Evaluation and Mitigation Strategy (REMS) program.31 Vandetanib is a CYP3A4 substrate, and the simultaneous use of strong CYP3A4 inducers should be avoided.
Cabozantinib is dosed at 140 mg orally once daily without food and is associated with gastrointestinal perforation and hemorrhage.33 Cabozantinib is not recommended in moderate to severe hepatic impairment (>1.5´ ULN) or in patients with severe renal impairment.33 Therapy should be withheld if grade 3 or greater hematologic and nonhematologic toxicities or intolerable grade 2 reactions occur. Cabozantinib is also a CYP3A4 substrate, and concomitant use with CYP3A4 inhibitors as well as inducers is not recommended.33
It is unclear whether both vandetanib and cabozantinib should be added to a formulary. Cabozantinib was evaluated in patients with progressive disease, but it was not a requirement for patients on vandetanib.31 Some patients who have failed vandetanib might be good candidates for cabozantinib; however, the inverse might not necessarily hold true due to lack of data.31 Patients who have a prolonged QT interval should more likely be treated with cabozantinib.31 Both drugs do not show an OS due to immature data.31 More research is needed to determine why some patients respond to some TKIs and not to others.31
Metastatic melanoma
Metastatic melanoma is a malignant tumor of the melanocytes and is associated with a poor prognosis. Early therapies used in the treatment of metastatic melanoma yielded low response rates, approximately 10% to 20%.34 Only a small fraction of these responses were considered complete response (CR).35 Approximately 40% to 60% of melanomas carry an activating mutation in BRAF which leads to downstream signaling.35,36 The aim of therapy is to improve OS, PFS, as well as to achieve a CR or partial response (PR). CR is defined as a complete disappearance of target lesions, and PR is a 30% decrease in diameter from baseline.35,36
Vemurafenib is an oral inhibitor of some mutated forms of BRAF serine-threonine kinase, including BRAFV600E.35 It is associated with improved OS and PFS (RR of death=0.37; RR of death or progression=0.26; P<0.001).36 Photosensitivity and cutaneous squamous cell carcinoma were the most common cutaneous AEs, and arthralgia was the most common noncutaneous AE reported with vemurafenib.36,37 Regular dermatologic evaluation with referral to a dermatologist is recommended.34 Patients should be evaluated at baseline and then every 2 months while on treatment.34 All patients are advised to avoid sun exposure in addition to wearing protective clothing and UVA/UVB sunscreen with a sun protection factor >30.34 Patients should also be monitored for other adverse reactions such as joint pain and swelling.36,37 Vemurafenib is recommended for patients with V600 mutation of the BRAF gene documented by an FDA-approved or Clinical Laboratory Improvement Amendments (CLIA)-approved facility.36,37 Another drug, dabrafenib, was approved on May 30, 2013, and is another oral inhibitor of BRAF. However, no information is forthcoming at this time.
Formulary considerations
Vemurafenib is administered at 960 mg orally twice daily with the first dose taken in the morning and the next approximately 12 hours later. It is a good choice for addition to the formulary since it is recommended by the NCCN Guidelines® as category 1.34 The other preferred regimen is ipilimumab and it is given intravenously.34 Approximately 90% of patients with the BRAF mutation have the V600 variant for which this medication has shown superior efficacy.36 Dose modification should be done in patients with symptomatic adverse drug reactions or those with prolonged QT. Treatment should be permanently discontinued in patients with a third appearance of an intolerable grade 2 to 3 and second appearance of a grade 4 AE. No adjustments are necessary in patients with cutaneous squamous cell carcinoma. This drug should be used with caution in patients who have severe hepatic or renal failure and is a CYP3A4 substrate, which should be used cautiously when administered with CYP3A4 inducers or inhibitors. Vemurafenib also is a moderate CYP1A2 inhibitor, weak CYP2D6 inhibitor, and a CYP3A4 inducer and should be used with caution simultaneously with medications metabolized by these enzymes.36,37
Pricing recommendations
Imatinib, dasatinib, nilotinib, and bosutinib are indicated for the primary treatment of chronic-phase adult CML. However, there are price variations among them ranging from $5,000 to $11,000 dollars per month with nilotinib costing the least and dasatinib the most (Table 2).38 Nilotinib is also given twice daily as opposed to the others which are given once daily. A comparison price table (Table 3) lists category 1 and 2A recommendations in advanced RCC and includes both the IV and oral formulations. Sunitinib and pazopanib are currently category 1 recommendations; however, there is a significant price difference of $5,000 making pazopanib more cost effective (Table 3).38 Comparison of cabozantinib and vandetanib is shown in Table 4.38 The monthly cost of cabozantinib is almost two and a half times the cost of vandetanib, making the latter more cost effective for patients. Vemurafenib’s monthly cost was compared to an older oral agent, temozolomide, and a newer IV agent, ipilimumab (Table 5).38 Temozolomide is almost half the cost while ipilimumab is almost 5 times more expensive than vemurafenib. Determining the agents most favorable for your formulary depends on physician preference, accessibility, cost, safety, and tolerability, in addition to primary endpoints like OS and PFS.
SUMMARY
Strategies proposed by Barnes et al can help equalize access for oral and IV medications.5 Changes should be made to the coverage and reimbursement system for oral oncolytics increasing prescribing incentives, thus ensuring physicians are adequately reimbursed.5 This would eventually lead to increased system efficiency and optimal patient access.5 The strategies they propose include creating a universal enrollment form for all patient assistance programs, streamlining administrative paperwork, moving all oral oncolytics under the medical benefit, establishing provider reimbursement for oncology treatment planning, and creating a specific oncology benefit.5
Table 1. Oral oncolytic agents FDA-approved since 2007
Year of approval
Oral oncolytic
FDA-approved indication
2007
Nilotinib (Tasigna)
Ph chromosome-positive chronic myelogenous leukemia
2007
Lapatinib (Tykerb)
HER2-positive advanced or metastatic breast cancer
2009
Pazopanib (Votrient)
Advanced renal cell carcinoma
2011
Crizotinib (Xalkori)
Locally advanced or metastatic anaplastic lymphoma kinase (ALK) −positive non−small-cell lung cancer
2011
Abiraterone acetate (Zytiga)
Metastatic castration-resistant prostate cancer
2012
Bosutinib (Bosulif)
Chronic, accelerated, or blast-phase Ph chromosome-positive chronic myelogenous leukemia
2012
Axitinib (Inlyta)
Advanced renal cell carcinoma
2012
Regorafenib (Stivarga)
Advanced colorectal cancer
2012
Enzalutamide (Xtandi)
Metastatic castration-resistant prostate cancer
2012
Cabozantinib (Cometriq)
Medullary thyroid cancer
2012
Vandetanib (Caprelsa)
Medullary thyroid cancer
2012
Vemurafenib (Zelboraf)
Metastatic melanoma
2013
Dabrafenib (Tafinlar)
Metastatic melanoma
Formulary/Source: www.fda.gov.
Table 2. Drug prices for chronic myelogenous leukemia
Drug
AWP unit price
AWP monthly cost
Nilotinib (Tasigna) (400 mg BID)
$85.82
$5,320.84
Bosutinib (Bosulif)
(500 mg QD)
$327.25
$10,144.75
Imatinib (Gleevec)
(400 mg QD)
$255.80
$7,929.80
Dasatinib (Sprycel)
(100 mg QD)
$343.29
$10,641.99
Monthly = 31 days.
Formulary/Source: Ref 38
Table 3. Drug prices for metastatic renal cell carcinoma
Drug
AWP unit price
AWP monthly cost
Temsirolimus (Torisel)
(25 mg weekly)
$1,620.65
$6,482.60
Sunitinib (Sutent)
(50 mg QD)
$461.21
$12,913.88
Pazopanib (Votrient)
(800 mg QD)
200-mg tablet
$71.30
$7,985.60
Sorafenib (Nexavar)
(400 mg BID)
$93.24
$5,221.44
Axitinib (Inlyta)
(5 mg BID)
$183.46
$4,673.76
Everolimus (Afinitor)
(10 mg QD)
$336.58
$9,424.24
Monthly = 28 days.
Formulary/Source: Ref 38
Table 4. Drug prices for medullary thyroid cancer
Drug
AWP unit price
AWP monthly cost
Cabozantinib (Cometriq)
(140 mg QD)
20-mg tablet
$141.43
$30,690.31
Vandetanib
(Caprelsa)
(300 mg QD)
$407.88
$12,644.28
Monthly = 31 days.
Formulary/Source: Ref 38
Table 5. Drug prices for metastatic melanoma
Drug
AWP unit price
AWP monthly cost
Vemurafenib (Zelboraf)
(960 mg BID)
240-mg tablet
$51.18
$11,464.32
Ipilimumab (Yervoy)
(3 mg/kg IV q3 weeks for 4 doses)
$30,693.60
$61,387.20
Temozolomide (Temodar)
(200 mg/m2 daily ´ 5 days)
350 mg = 250-mg + 100-mg tablets
$911.24
$4,556.20
Monthly = 28 days.
Formulary/Source: Ref 38
References
1. Campbell M, Kaufman MB, Bendix J. Oral oncology drugs: navigating dispensing, billing, and reimbursement challenges is a daunting but not insurmountable task. Drug Topics. Feb 1., 2009. Available at: http://drugtopics.modernmedicine.com/news/oral-oncology-drugs. Accessed March 4, 2013.
2. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force report: oral chemotherapy. JNCCN. 2008;6(Suppl 3):S1–S14.
3. Goldman DP, Joyce GF, Lawless G, Crown WH, Willey V. Benefit design and specialty drug use. Health Aff (Millwood). 2006;25:1319−1331.
4. Streeter SB, Schwartzberg L, Husain N, Johnsrud M. Patient and plan characteristics affecting abandonment of oral oncolytic prescriptions. J Oncol Pract. 2011;7(3 Suppl):46s–51s.
5. Barnes L, Burich M, Little A, Nowak M, Haroldson B. Oral oncolytics/addressing the barriers to access and identifying areas for engagement. Executive summary. Washington, DC: Avalere Health, Feb. 2010. Available at: http://www.avalerehealth.net/wm/show.php?c=&id=842. Accessed March 4, 2013.
6. Henderson, L. Oncologists report declining reimbursement most significant challenge for future practice viability. KJT Group. February 12, 2009. Available at: http://kjtgroup.com/2009/02/oncologists-report-declining-reimbursement-most-significant-challenge-for-future-practice-viability/ Accessed February 18, 2013.
7. Liu G, Franssen E, Fitch M, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol. 1997;15:110−115.
8. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Chronic Myelogenous Leukemia. V.4.2013. © National Comprehensive Cancer Network, Inc. 2013. All rights reserved. Accessed March 1, 2013. To view the most recent and complete version of the guideline, go online to www.nccn.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
9. Faderl S, Talpaz M, Estrov Z, Kantarjian H. Chronic myelogenous leukemia: biology and therapy. Ann Intern Med. 1999;131:207−219.
10. O’Brien SG, Guilhot F, Larson RA, et al, for the IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994−1004.
11. Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28−37.
12. Jabbour E, Cortes J, Giles F, O’Brien S, Kantarjian H. Current and emerging treatment options in chronic myeloid leukemia. Cancer. 2007;109:2171−2181.
13. Guilhot F, Druker B, Larson R, et al. High rates of durable response are achieved with imatinib after treatment with interferon alpha plus cytarabine: results from the International Randomized Study of Interferon and STI571 (IRIS) trial. Haematologica. 2009;94:1669−1675.
14. Shah N, Tran C, Lee F, Chen P, Norris D, Sawyers C. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399−401.
15. Shah NP, Kim D-W, Kantarjian H, et al. Potent, transient inhibition of BCR-ABL with dasatanib 100 mg daily achieves rapid and durable cytogenetic responses and high transformation-free survival rates in chronic phase chronic myeloid leukemia patients with resistance, suboptimal response or intolerance to imatinib. Haematologica. 2010;95:232−240.
16. Kantarjian H, Flinn IW, Goldberg S, et al. Nilotinib versus imatinib in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): ENESTnd 3-year (yr) follow-up (f/u) [abstract]. J Clin Oncol. 2012;30(suppl):6509.
17. Khoury H, Cortes J, Kantarjian H, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119:3403−3412.
18. Cortes J, Kantarjian H, Bruemmendorf T, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118:4567-4576.
19. Cortes JE, Kim D-W, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30:3486−3492.
20. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Kidney Cancer. V.1.2013. © National Comprehensive Cancer Network, Inc. 2013. All rights reserved. Accessed March 1, 2013. To view the most recent and complete version of the guideline, go online to www.nccn.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
21. Motzer R, Hutson T, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115−124.
22. Motzer R, Hutson T, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584−3590.
23. Sternberg C, Davis I, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061−1068.
24. Sonpavde G, Hutson T, Rini B. Axitinib for renal cell carcinoma. Expert Opin Investig Drugs. 2008;17:741−748.
25. Rini B, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931−1939.
26. Keisner SV, Shah SR. Pazopanib: the newest tyrosine kinase inhibitor for the treatment of advanced or metastatic renal cell carcinoma. Drugs. 2011;71:443−454.
27. Motzer R, Hudson T, Reeves J, et al. Randomized, open-label, phase III trial of pazopanib versus sunitinib in first-line treatment of patients with metastatic renal cell carcinoma (mRCC): results of the COMPARZ trial [abstract]. Presented at the 2012 European Society for Medical Oncology; Vienna, Austria; October 1, 2012. Abstract LBA 8.
28. Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R D. 2011;11:113−126.
29. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Medullary Thyroid Carcinoma V.1.2013. © National Comprehensive Cancer Network, Inc. 2013. All rights reserved. Accessed March 1, 2013. To view the most recent and complete version of the guideline, go online to www.nccn.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
30. Wells S, Robinson B, Gagel R, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141.
31. Nagilla M, Brown RL, Cohen EE. Cabozantinib for the treatment of advanced medullary thyroid cancer. Adv Ther. 2012;29:925–934.
32. Schoffski P, Elisei R, Muller S, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline [abstract]. J Clin Oncol. 2012;30(Suppl 15):Abstract 5508.
33. COMETRIQ® (cabozantinib)[package insert]. South San Francisco, CA: Exelixis, Inc.; 2012. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/203756Orig1s000ltr.pdf. Accessed March 4, 2013.
34. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Melanoma V.2.2013. © National Comprehensive Cancer Network, Inc. 2013. All rights reserved. Accessed March 1, 2013. To view the most recent and complete version of the guideline, go online to www.nccn.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809−819.
36. Sosman J, Kim K, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707−714.
37.Chapman P, Hauschild A, Robert C, et al, for the BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507−2516.
38. Thomson Reuters Micromedex Clinical Evidence Solutions. Thomson Reuters; ©2011. RED BOOK Drug References; ©2011 [cited Feb 13, 2013]. Available at: http://thomsonreuters.com/products_services/healthcare/healthcare_products/clinical_deci_support/micromedex_clinical_evidence_sols/med_safety_solutions/red_book/. Accessed February 13, 2013.
Dr Bwayo Weaver is associate professor of pharmacy; Dr Moore is a pharmacy oncology fellow; Mr Shah is a candidate for doctor of pharmacy; and Dr Serlemitsos-Day is assistant professor of pharmacy, Howard University College of Pharmacy, Washington, D.C.
The authors report no financial disclosures as related to products discussed in this article.
Acknowledgments
We thank Dr. Fredric Lombardo and Dr. Rosemary Olajide for helping with the proofreading and editing of this manuscript.






Payers Recognize the Benefits, but Still See Weight Loss Drugs through a Cost Lens
April 12th 2024Jeffrey Casberg, M.S., R.Ph., a senior vice president of clinical pharmacy at IPD Analytics LLC, a drug intelligence firm that advises payers and pharmaceutical companies, talks about how payers are thinking about weight-loss drugs.

