
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Pulmonary arterial hypertension: Bridging the gap between efficacy, quality of life, and cost-effectiveness
Pulmonary arterial hypertension (PAH) is a rare but progressive condition characterized by abnormal proliferation and remodeling, vasoconstriction, and thrombosis of the pulmonary vasculature, leading to elevated pulmonary arterial pressure, increases in peripheral vascular resistance, and ultimately to right heart failure and death. Recently, the therapeutic armamentarium for PAH has expanded.



Key Points
Abstract
Pulmonary arterial hypertension (PAH) is a rare but progressive condition characterized by abnormal proliferation and remodeling, vasoconstriction, and thrombosis of the pulmonary vasculature, leading to elevated pulmonary arterial pressure, increases in peripheral vascular resistance, and ultimately to right heart failure and death. Recently, the therapeutic armamentarium for PAH has expanded. With the significant costs associated with most of these agents as well as its complications, managed care organizations struggle with determining the most appropriate and cost-effective management of PAH. A cost-effective evidence-based treatment algorithm is proposed with guidance from clinical data and reviews from the literature that assess disparites in clinical improvement, quality of life, rates of mortality, and total healthcare costs between agents. The algorithm constructs a hierarchy of therapies based on acute vasoreactivity testing, and suggests the use of high-dose oral calcium channel blockers as a first option for patients with a positive response to testing, phosphodiesterase-5 inhibitor monotherapy as first-line treatment for lower risk (functional class [FC] II or III based on World Health Organization (WHO)/New York Heart Association (NYHA) classification system) patients with a negative response to testing, and prostacyclins as first-line agents for higher risk (WHO/NYHA FC IV) patients with a negative response to testing. (Formulary. 2010;45;190–199.)


CLASSIFICATION OF PAH

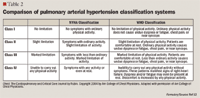
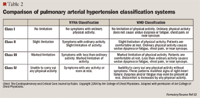
Recent data suggest differences in the classification of patients when classified using one system versus the other (NYHA vs WHO).11 An assessment of interrater reliability between the NYHA and WHO functional classification systems (applied by clinicians to 10 hypothetical patients) found wide variation in assignment of functional class (FC) between the two systems. Furthermore, clinicians' rankings of the patients' functional classification spanned 3 functional classes across virtually all patients. This study effectively demonstrated the inconsistency in classification ranking between clinicians. The lack of interrater reliability in assignment of functional class can have a significant impact on management decisions, treatment outcomes, and ultimately the costs associated with their treatment.
PHARMACOLOGIC AGENTS
The current pharmacotherapeutic approach to PAH focuses on 4 primary classes of agents: calcium channel blockers (CCBs), phosphodiesterase-5 (PDE-5) inhibitors, endothelin receptor antagonists (ERAs), and prostacyclins. All agents demonstrate vasodilatory and antiproliferative effects in the pulmonary vasculature with the exception of CCBs.14,15 The American College of Cardiology Foundation/American Heart Association Expert Consensus Document (ACCF/AHA) recommends conventional therapies (anticoagulation, diuretics, oxygen, and digoxin) for the treatment of comorbidities associated with PAH.2
Calcium channel blockers. Calcium channel blockers exhibit vasodilatiory effects in the smooth muscle cells of the pulmonary arteriolar system.13,14,16 The efficacy of CCB therapy for PAH was first suggested in 1992 in a small subset of patients that responded with reductions in pulmonary artery pressure and pulmonary vascular resistance to high doses.16 A retrospective analysis of 557 patients determined that only 6.8% of patients were long-term CCB responders.17 Despite the low long-term response, high-dose CCBs are still considered a powerful option in patients who respond positively to acute vasoreactivity testing, and their use should be limited to this setting.1,2 CCBs do not possess antiproliferative effects, which may explain why most patients do not respond to CCB therapy.15 If a patient does not improve to FC I or II after 3 months of therapy with CCBs, the patient should be considered a nonresponder and adjunctive medications or other altogether different therapies should be considered.2 If a CCB is the chosen therapy for PAH, guidelines from the American College of Chest Physicians (ACCP) recommend the use of long-acting nifedipine, diltiazem, or amlodipine, and avoidance of verapamil.1
Phosphodiesterase type-5 inhibitors. Phosphodiesterase type-5 (PDE-5) inhibitors enhance nitric oxide-dependent, cyclic guanosine monophosphate (cGMP)-mediated pulmonary vasodilation and antiproliferation of vascular smooth muscle.8,14 Two FDA-approved PDE-5 inhibitors are currently indicated for use in PAH: sildenafil and tadalafil.
SILDENAFIL
In a randomized, double-blind, controlled trial, 278 patients with PAH and idiopathic PAH (WHO FC II–IV) were randomly assigned to sildenafil 3 times daily or placebo for 12 weeks. The change in 6-minute walk distance (6MWD) from baseline, the primary end point of the study, increased an average of 13.7% (47 m, P<.001 for all comparisons) across all doses (20 mg, 40 mg, 80 mg). After 12 weeks, WHO FC improved by at least 1 FC in 21% (P=.003) of patients randomly assigned to 20 mg, 29% (P<.001) in those randomly assigned to 40 mg, and 35% (P<.001) of patients randomly assigned to 80 mg. In the 222 patients who completed 1 year of treatment with sildenafil, improvement from baseline in 6MWD was 17.6% (an increase of 51 m). Long-term improvement with sildenafil is based upon patients who received up to 80 mg 3 times daily, whereas the FDA-approved dosage is 20 mg 3 times daily.1,8
TADALAFIL
Tadalafil, the most recent PDE-5 inhibitor approved, was studied in a 16-week, double-blind, placebo-controlled trial, in 405 patients with PAH in WHO FC II or III (treatment-naïve or currently taking bosentan) in which patients were randomly assigned to placebo or 1 of 4 daily doses of tadalafil (2.5 mg, 10 mg, 20 mg, or 40 mg). Tadalafil, 40 mg once daily, significantly improved 6MWD (P<.01), the primary end point, in a dose-dependent manner by week 16 of the study. Tadalafil, 40 mg once daily, improved 6MWD by 33 m and was the only treatment arm that achieved statistical significance. The improvement in 6MWD was greater in treatment-naïve patients (44 m, P<.01) compared with those on background bosentan therapy (23 m, P=.09). The blunted response observed with bosentan coadministration may be explained by reduced tadalafil plasma levels due to cytochrome P450 3A4 interaction, which potentially decreases the pharmacodynamic effect of tadalafil. Tadalafil, 40 mg, appeared to be the optimal dose for PAH patients to improve exercise capacity, clinical worsening, QoL, and hemodynamic measures.18
ERAS
Serum levels of endothelin-1, an endogenous vasoconstricter that encourages proliferation of smooth muscle in the pulmonary vasculature, are elevated in patients with PAH. Endothelin receptor antagonists (ERAs) inhibit receptors of endothelin-1, ETA ± ETB, to result in vasodilation and antiproliferation of the pulmonary vasculature.13 There are currently 2 ERAs approved in the United States; bosentan (nonselective) and ambrisentan (ETA selective blocker).
Bosentan. Bosentan has been evaluated by several groups for its effect in PAH. In BREATHE-1, a double-blind, multicenter, placebo-controlled study, 213 patients with PAH, in WHO FC III (90% of patients) or IV, were randomly assigned to placebo or bosentan, 62.5 mg twice daily, for 4 weeks followed by either 125 mg or 250 mg twice daily for at least 12 weeks.19 After 16 weeks of treatment, the primary end point of 6MWD increased by a mean of 36 m in the bosentan groups compared to a decrease of 8 m in the placebo group (P<.001). In comparing the 125-mg and 250-mg twice-daily doses, the placebo-corrected improvement was more pronounced for the 250-mg twice-daily dose (54 m). By week 16, 38% of patients assigned to 125 mg and and 34% assigned to 250 mg of bosentan improved to WHO FC II. Furthermore, bosentan treatment was found to significantly increase time to clinical worsening (secondary end point), compared with placebo (P=.002).
Ambrisentan. Ambrisentan is an oral, once-daily, selective ERA approved by FDA for the treatment of PAH (WHO FC II and III). Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter Efficacy Study (ARIES) 1 and 2 were concurrent, double-blind, placebo-controlled, randomized trials.20 In ARIES-1 and ARIES-2, 202 and 192 patients with PAH (WHO FC I-IV) received 5 mg or 10 mg, and 2.5 mg or 5 mg ambrisentan for 12 weeks, respectively. The 6MWD from baseline (primary end point for both studies) improved in all ambrisentan groups in both trials. The placebo-corrected treatment effects were improvements of 31 m (P=.008) and 51 m (P<.001) in the 5-mg and 10-mg ambrisentan groups, and 32 m (P<.022) and 59 m (P<.001) in the 2.5-mg and 5-mg ambrisentan groups, respectively. In the 280 patients who completed 48 weeks of ambrisentan monotherapy, 6MWD improved by an average of 39 m.
A 2009 Cochrane Collaboration review in which the efficacy of ERAs in PAH was evaluated from 11 randomized controlled trials involving 1,457 patients, concluded that ERAs improve WHO/NYHA FC status, reduce dyspnea, improve cardiopulmonary variables, and improve 6MWD in patients with PAH with WHO/NYHA FC II and III.21
Prostacyclins. Prostacyclin induces relaxation of vascular smooth muscle, inhibits the proliferation of smooth muscle cells, and is a powerful inhibitor of platelet aggregation.13 Prostacyclin synthase is reduced in PAH patients, which ultimately results in decreased production of prostacyclin.2 Prostacyclin supplementation has long been considered the gold standard of therapy by many clinicians. Currently, 3 prostanoids are approved by FDA for use in patients with PAH: epoprostenol, treprostinil, and iloprost.
Epoprostenol. The use of epoprostenol in the treatment of PAH dates back to the early 1980s.22 For years, it has been regarded as the most effective medication to treat advanced stages of PAH. Epoprostenol must be administered via intravenous infusion due to its half life of <6 minutes.23 Many open-label and randomized controlled trials have demonstrated improvements in 6MWD, FC, and hemodynamics with epoprostenol treatment.24-27 Positive long-term survival data have also been reported with intravenous epoprostenol.26,27 According to the American College of Chest Physician guidelines, epoprostenol should be reserved for patients diagnosed with PAH WHO FC III or IV only because of complications, such as sepsis, from having a central line for continuous infusion.1
Treprostinil. Treprostinil is a longer-acting prostacyclin that is available in subcutaneous, intravenous, and inhaled forms. In a randomized trial of 470 patients with PAH (NYHA FC II, III, and IV) who were assigned to placebo or subcutaneous treprostinil, a placebo-corrected improvement in 6MWD of a median of 16 m (P=.006) occurred in treprostinil-treated patients.28 Subcutaneous infusion was associated with injection site reactions such as pain and erythema in 85% of patients. In an analysis of 860 patients to determine long-term outcomes in PAH patients treated with subcutaneous treprostinil monotherapy, survival ranged from 87% to 68% over 1 to 4 years.29 In a 12-week open-label study of 27 patients with PAH (WHO FC II and III), no statistically significant difference was observed in 6MWD after transitioning patients from intravenous epoprostenol to intravenous treprostinil.30
Treprostinil has recently received FDA approval as an inhaled formulation. Inhaled treprostinil was studied in a 12-week, randomized, double-blind, placebo-controlled multicenter study called TRIUMPH-I in which 235 patients with PAH were enrolled. Patients had predominantly NYHA FC III symptoms and were receiving either background bosentan (70%) or background sildenafil (30%) for at least 3 months, and may also have been on CCBs or conventional therapy.31 Patients were randomly assigned to placebo or inhaled treprostinil in 4 daily treatment sessions, titrated to 9 breaths per session. By week 12, patients randomly assigned to inhaled treprostinil had a placebo-corrected improvement of 20 m (P<.001) in 6MWD from baseline.
Iloprost. Inhaled iloprost is indicated for the treatment of PAH in patients with NYHA FC III or IV symptoms. In a randomized, double-blind, multicenter, placebo-controlled trial of 203 patients with NYHA FC III (59%) or IV PAH, inhaled iloprost or placebo was added to the patients' current therapy (CCBs or conventional therapy) with no adjunctive prostacyclins or ERAs.32 Iloprost was shown to improve 6MWD at week 12 by at least 10% in 17% of patients compared to 5% of patients receiving placebo (P=.007).
The study also showed improved 6MWD by a placebo-corrected distance of 40 m and decreased clinical deterioration. In a multicenter, double-blind trial, 67 NYHA FC III patients who were stable for 12 weeks while on bosentan treatment were randomly assigned to placebo or inhaled iloprost.33 At week 12, 34% of patients receiving iloprost improved by 1 FC compared with 6% in the placebo group (P=.002). Iloprost was also found to delay time to clinical worsening (P=.0219). The iloprost group used an average of 5.6 inhalations per day and the average total daily dose was 27.8 μg (range, 11.6–33.3 μg).33
END POINTS, CLINICAL IMPLICATIONS
Various end points have been used historically to evaluate clinical improvement or the regression of symptoms attributable to drug therapy. How accurately these measures quantify and differentiate between a clinically meaningful improvement in long-term outcomes versus a QoL improvement is a subject of debate. In order for these measures to be useful, they must be easily reproducible and affordable in all practice settings, and consistently reflect the impact of drug therapy on disease state progression or improvement.
Hemodynamics, plasma biomarkers, FC. Improvements in hemodynamic measurements are not reliable surrogate end points because such improvements do not translate to improved survival and show practically no correlation to long-term outcomes.
Although promising as surrogate end points, little data have been collected on the use of plasma biomarkers and cardiac imaging and outcomes. Further, they can be time consuming, costly, and technically infeasible.34 Although a large body of literature supports the use of improvement of FC as an outcomes end point, this measure is extremely subjective.35
6MWD. Assessment of exercise capacity in the form of 6MWD is the most commonly studied primary end point in clinical trials, and many have found a correlation of this measure with mortality. It is reproducible, inexpensive, and relatively reliable. 34 Its use in discerning a clinically significant change in NYHA FC II patients is limited, however.36 Additionally, the level of improvement over baseline that constitutes a minimally important difference is just now being addressed. Gilbert et al suggest that 41 m over baseline 6MWD is a minimally important difference that is a statistically significant change in outcome.37 Whether this difference translates into improvements in long-term outcomes (eg, survival) has yet to be defined.
Combined end points. Combined end points such as "time to clinical worsening" incorporate multiple end points that may include death, hospitalization, need for additional therapy, and lung transplantation. They offer a more global assessment of disease state with increased statistical power and are now being explored. The clinical significance may be debatable as combined end points suffer from confounders that erroneously dilute and/or compound events.34
META-ANALYSIS, END POINT CORRELATION TO SURVIVAL
Three meta-analyses of randomized, controlled trials have attempted to determine the likelihood of a significant survival benefit with the different PAH agents, and came to varied conclusions.38-40
An inference can be made from these meta-analyses that current study designs are unable to correlate 6MWD to an increase in survival. Once a minimally important difference in an end point is defined and quantitatively connected to long-term outcomes, the logical progression is to ask if the treatment positively impacts patient QoL and at what cost to society.
QUALITY OF LIFE AND 6MWD
Quality-of-life measures are critical in the evaluation of PAH because changes in physiologic measures do not necessarily reflect tangible benefits as perceived by the patient. Various instruments have been used in clinical studies as secondary end points, including the Medical Outcome Study: 36-item Short Form Health Survey (SF-36) to measure QoL. Statistically significant differences in QoL have been reported by a number of investigators using a variety of agents.18,24,28,41-46 Benchmarks are still needed in order to evaluate these findings and place them within a context to determine if a minimum threshold has been reached that represents an important change or benefit. Domains assessed and scoring methods used differ between these tools, and often the data reported from clinical trials is poor and incomplete, making comparisons of the results difficult. Additionally, they are not specific to PAH. A PAH-specific tool, the Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR), was recently developed. It has not yet been broadly implemented in clinical trials. A clear connection between QoL measures and physiologic measures such as 6MWD is important in evaluating both clinical improvement and benefit to the patient.47
The evaluation of the QoL benefits achieved by pharmacologic treatments, as opposed to exercise training alone, has yet to be addressed. A small randomized, controlled study of patients with WHO FC II-IV found that exercise training alone significantly improved SF-36 scores across all domains, and increased 6MWD by 111 m over baseline.46 This comparison of PAH agents to exercise training alone may prove to be an important piece of information when cost in relation to effectiveness enters into the decision-making model.
PHARMACOECONOMIC (PE) STUDIES
Cost minimization analysis and cost effectiveness analysis, with cost utility analysis (CUA) that measures effectiveness in quality adjusted life years (QALYs), would allow a comparison of the costs of treatment to health-related outcomes. The lack of existing PE analysis of PAH drug therapy limits our ability to make fully informed decisions concerning therapy.
Treprostinil, Epoprostenol, Iloprost. Narine et al and Einarson et al evaluated healthcare costs in a comparison of treprostinil and epoprostenol.48,49 Treprostinil yielded a savings of $12,478 USD and $14,504 CAD (~$14,149 USD) per patient per year, respectively over epoprostenol.47 Einarson attributed the cost savings with treprostinil to a decrease in hospitalizations and adverse drug events.48 One major limitation of these studies was the assumption of equipotent dosing. In a 2006 study, treprostinil was found to be exponentially more expensive (2-3 fold) after adjusting for equipotent dose to epoprostenol.9 Epoprostenol (30 to 39 ng/kg/min) was determined to cost $48,027 to $63,620 USD per year, whereas treprostinil (60 to 69 ng/kg/min) was determined to exceed $142,350 USD per year in costs.9 A CUA of iloprost by its manufacturer found it was both less costly and more effective than epoprostenol.47 The study assumed a fixed cost for all doses of iloprost. A CUA using a Markov model found strikingly dissimilar results. Iloprost, compared to treprostinil or epoprostenol, resulted in a small gain of 0.08 QALYs per 100 patients, but at a significant cost per patient year of $382,310 USD (vs treprostinil) and $144,348 USD per patient year (vs epoprostenol) per QALY.3
Bosentan and sildenafil. A CUA by Highland in the United States in 2003 found that bosentan was less costly and resulted in a greater number of QALYs (11 QALYs per 100 patients at a savings of $36,319 USD per patient year) than either treprostinil or epoprostenol.50 In 2006, Wlodarczk in Australia analyzed bosentan versus conventional therapy (CCBs and oxygen were defined as conventional therapy) and found an incremental cost effectiveness ratio of $55,927 USD per life year gained with bosentan.51 Each QALY cost £22,058 (approximately $33,616 USD) with sildenafil, which was less costly compared with prostacyclins and ERAs.47
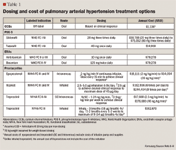
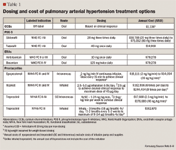
PROPOSED TREATMENT ALGORITHM

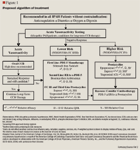
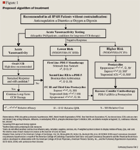
Among the second-line choices (ERAs) for patients in FC II or III, bosentan and ambrisentan are similar in efficacy and QoL, while ambrisentan lacks PE data. The lack of cost-effectiveness data of ambrisentan is the main reason we have placed bosentan as the preferred second-line option. Although ambrisentan is associated with fewer drug-drug interactions than bosentan, the interactions with bosentan can be easily managed.
We do not recommend the use of prostacyclins in IPAH patients presenting with FC II symptoms. The cost and complexity of medication administration associated with epoprostenol, treprostinil, and iloprost place these agents third-line in FC III patients. Furthermore, aside from subcutaneous treprostinil, none of the prostacyclin agents are indicated for use in FC II patients.
We recommend PDE-5 and ERAs in FC III patients as first and second-line agents respectively and inhaled treprostinil and iloprost as third-line agents based on efficacy, adverse effect profile, and cost. In patients with FC IV, epoprostenol is favored over iloprost given its better efficacy and cost profile, although the risk of complications is higher with epoprostenol.
The most optimal combination therapy and point of initiation is not clearly defined at this time. The utility of combination therapy trials to date is limited by a number of confounders such as lack of power due to small sample size, retrospective design, and debatable benefit. Patients enrolled in most of these studies were primarily unresponsive to monotherapy or deteriorated while receiving monotherapy; therefore, whether the benefit seen was the result of the new agent alone or synergistic effect of the combination is debatable.52
CONCLUSION
Considering the overall lack of interrater agreement in FC, emphasis should be placed upon accurate diagnosis and classification of PAH, as incorrect classification can lead to inappropriate treatment, increased risk to patients, and unnecessary utilization. In addition, given that treatment options have increased, earlier diagnosis should be emphasized so that patients can be treated before presenting with advanced stage of the disease. Although efficacy and clinical appropriateness are the most important variables to consider, it should not be the sole focus when designing a pharmacologic treatment plan for patients with PAH. Balancing efficacy with QoL and costs should be considered. Future head-to-head trials of agents within classes and agents between classes, are needed to effectively guide clinical practice. Trials addressing health-related QoL and PE considerations as well as optimal combination approaches are needed to establish more comprehensive clinical practice guidelines. Furthermore, a need to adopt new trial designs that can address the current issue of clinical relevance of primary and secondary end points is long overdue.
Dr Gandhi is a managed care pharmacy practice resident, Health Plan of San Joaquin, French Camp, Calif. Mr Baker is a PharmD candidate at the University of the Pacific, Stockton, Calif. Dr Shek is an associate professor of pharmacy practice, University of the Pacific, and director of pharmacy, Health Plan of San Joaquin. Dr Yeh is a disease management clinical pharmacist, Health Plan of San Joaquin. Dr Bishop is medical director, Health Plan of San Joaquin.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917–1928.
2. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–1619.
3. Garin MC, Clark, L, Chumney E, Simpson KN, Highland KB. Cost-utility of treatments for pulmonary arterial hypertension. A Markov State Transition Decision Analysis Model. Clin Drug Investig. 2009;29:635–646.
4. Hoeper MM. Definition, classification, and epidemiology of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:369–375.
5. Hyduk A, Croft JB, Ayala C, Zheng K, Zheng Z-J, Measah GA. Pulmonary hypertension surveillance - United States, 1980-2002. MMWR Surveill Summ. 2005;54:1–28.
6. Facts & Comparisons. Available at: http://www.factsandcomparisons.com/. Accessed on March 7, 2010.
7. First Data Bank. Available at: http://www.firstdatabank.com/. Accessed on March 22, 2010
8. Galiè N, Ghofrani HA, Toribicki A, et al; Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005; 353:2148–2157.
9. Oudiz RJ, Farber HW. Dosing considerations in the use of intravenous prostanoids in pulmonary arterial hypertension: an experience-based review. Am Heart J. 2009;157:625–635.
10. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431.
11. Taichman DB, McGoon MD, Harhay MO, et al. Wide variation in clinicians' assessment of New York Heart Association/World Health Organization functional class in patients with pulmonary arterial hypertension. Mayo Clin Proc. 2009;84:586–592.
12. Rubin LJ. Introduction: diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:7S–10S.
13. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665.
14. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436.
15. Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;5:1527–1538.
16. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81.
17. Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–3111.
18. Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–2903.
19. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903.
20. Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the Ambrisentan in Pulmonary Arterial Hypertension, Double-Blind, Placebo-Controlled, Multicenter, Efficacy (ARIES) Randomized, study 1 and 2. Circulation. 2008;117:3010–3019.
21. Liu C, Chen J, Gao Y, Deng B, Liu K. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database of Systematic Reviews. 2009;Issue 3, Art. No.:CD004434.
22. Higenbottam T, Wheeldon D, Wells F, et al. Long-term treatment of primary pulmonary hypertension with continuous intravenous epoprostenol (prostacyclin). Lancet. 1984;1:1046–1047.
23. Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112:485–491.
24. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension: the Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996; 334:296–302.
25. Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to scleroderma spectrum of disease: a randomized controlled trial. Ann Intern Med. 2000;132:425–434.
26. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40:780–788.
27. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477–1482.
28. Simonneau G, Barst RJ, Galiè N, et al; Treprostinil Study Group. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arerial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–804.
29. Barst RJ, Galiè N, Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195–1203.
30. Gomberg-Maitland M,Tapson VF, Benza R, et al. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med. 2005;172:1586–1589.
31. Tyvaso [package insert]. Research Triangle Park, NC: United Therapeutics Corp.; 2009.
32. Ventavis [package insert]. South San Francisco, CA: Acetelion Pharmaceuticals US, Inc.; 2009.
33. McLaughlin VV, Oudiz RJ, Frost A. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174;1257–1263.
34. Ventetuolo CE, Benza R, Peacock AJ, Zamanian RT, Badesch DB, Kawut SM. Surrogate and combined endpoints in pulmonary arterial hypertension. Proc Am Thorac Soc. 2008;5:617–622.
35. McLaughlin VV, Badesch DB, Delcroix M, et al. Endpoints and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S97–107.
36. Gomberg-Maitland M, Huo D, Benza RL, et al. Creation of a model comparing 6-minute walk test to metabolic equivalent in evaluating treatment effects in pulmonary arterial hypertension. J Heart Lung Transplant. 2007;26:732–738.
37. Gilbert C, Brown M, Cappelleri J, Carlsson M, McKenna SP. Estimating a minimally important difference in pulmonary arterial hypertension following treatment with sildenafil. Chest. 2009;135:137–142.
38. Macchia A, Marchioli R, Marfisi R, et al. A meta analysis of trials of pulmonary hypertension: A clinical condition looking for drugs and research methodology. Am Heart J. 2007;153:1037–1047.
39. Gailè N, Manes A, Negro L, Palazzini M, Bacchi-Raggioni ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30:394-403.
40. Macchia A, Marchiloi R, Tognoni G, et al. Systematic review of trials using vasodilators in pulmonary arterial hypertension: why a new approach is needed. Am Heart J. 2010;159:245–257.
41. Wilkins MR, Paul GA, Strange JW, et al. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171:1292–1297.
42. Sastry BK, Narasimhan C, Reddy NK, Raju S. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo controlled, double blinded, crossover study. J Am Coll Cardiol. 2004;43:1149–1153.
43. Keogh AM, McNeil KD, Wlodarczyk J, et al. Quality of life in pulmonary arterial hypertension: improvement and maintenance with bosentan. J Heart Lung Transplant. 2007;26:181–187.
44. Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46:529–535.
45. Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–329.
46. Mereles D, Ehlken N, Kreuscher S, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation. 2006;114:1482–1489.
47. Chen YF, Jowett S, Barton P, et al. Clinical and cost-effectiveness of epoprostenol, iloprost, bosentan, sitaxentan, and sildenafil for pulmonary arterial hypertension within their licensed indications: a systemic review and economic evaluation. Health Technol Assess. 2009;13:1–320.
48. Narine L, Hague LK, Walker JH, et al. Cost-minimization analysis of treprostinil vs. epoprostenol as an alternate to oral therapy non-responders for the treatment of pulmonary arterial hypertension. Curr Med Res Opin. 2005;21:2007–2016.
49. Einarson TR, Granton JT, Vicente C, Walker JH, Engel G, Iskedjian M. Cost-effectiveness of treprostinil versus epoprostenol in patients with pulmonary arterial hypertension: a Canadian analysis. Can Respir J. 2005;12:419–425.
50. Highland K, Strange C, Mazur J, et al. Treatment of pulmonary arterial hypertension. A preliminary decision analysis. Chest. 2003;124:2087–2092.
51. Wlodarczyk JH, Cleland LG, Keogh AM, et al. Public funding of bosentan for the treatment of pulmonary artery hypertension in Australia. Cost effectiveness and risk sharing. Pharmacoeconomics. 2006;24(9):903–915.
52. Abraham T, Wu G, Vastey F, Rapp MS, Saad N, Balmir E. Role of combination therapy in the treatment of pulmonary arterial hypertension. Pharmacotherapy. 2010;30:390–404.


Coalition promotes important acetaminophen dosing reminders
November 18th 2014It may come as a surprise that each year Americans catch approximately 1 billion colds, and the Centers for Disease Control and Prevention estimates that as many as 20% get the flu. This cold and flu season, 7 in 10 patients will reach for an over-the-counter (OTC) medicine to treat their coughs, stuffy noses, and sniffles. It’s an important time of the year to remind patients to double check their medicine labels so they don’t double up on medicines containing acetaminophen.
Support consumer access to specialty medications through value-based insurance design
June 30th 2014The driving force behind consumer cost-sharing provisions for specialty medications is the acquisition cost and not clinical value. This appears to be true for almost all public and private health plans, says a new report from researchers at the University of Michigan Center for Value-Based Insurance Design (V-BID Center) and the National Pharmaceutical Council (NPC).
Management of antipsychotic medication polypharmacy
June 13th 2013Within our healthcare-driven society, the increase in the identification and diagnosis of mental illnesses has led to a proportional increase in the prescribing of psychotropic medications. The prevalence of mental illnesses and subsequent treatment approaches may employ monotherapy as first-line treatment, but in many cases the use of combination of therapy can occur, leading to polypharmacy.1 Polypharmacy can be defined in several ways but it generally recognized as the use of multiple medications by one patient and the most common definition is the concurrent use of five more medications. The presence of polyharmacy has the potential to contribute to non-compliance, drug-drug interactions, medication errors, adverse events, or poor quality of life.
Medical innovation improves outcomes
June 12th 2013I have been diagnosed with stage 4 cancer of the pancreas, a disease that’s long been considered not just incurable, but almost impossible to treat-a recalcitrant disease that some practitioners feel has given oncology a bad name. I was told my life would be measured in weeks.

