
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Specialty pharmacy's role in REMS, FDA's new drug safety program
Specialty pharmacies are uniquely positioned to play a key role in the implementation of Risk Evaluation and Mitigation Strategies (REMS).



Key Points
Abstract
Prescription drug safety is a topic that generates a great deal of attention in the headlines, among patient communities, and in the US civil court system. A problem arises when a drug offers significant therapeutic benefits but also carries challenging health or safety risks. FDA recently introduced the Risk Evaluation and Mitigation Strategy (REMS) to address this dilemma. Many of the newest approved therapies are biologics and are subject to REMS, which can call for communications development, patient registries, ongoing patient monitoring, and certification for prescribers and pharmacies. Specialty pharmacies are uniquely positioned to play a key role in providing patients access to these drugs, given their high level of patient and physician interaction, specially trained therapy teams, and experience with high-intensity therapies. (Formulary. 2009;44:300–308.)



REMS may be mandated for a newly approved drug that has a known safety risk or for a drug already on the market that has a safety risk not previously identified during clinical trials. Each REMS program includes requirements that go beyond product labeling, as well as a timetable to assess the program's effectiveness in meeting stated goals.
Despite providing patients with new therapeutic options for debilitating or life-threatening conditions, many biologics require a degree of special management to monitor potential health and safety risks. Without a REMS program, many biologic drugs might not reach introduction into the market. "These safety plans allow patients to have access to certain medicines for which there are safety concerns that can be managed through appropriate use," said Jane Axelrad, associate director for policy, Center for Drug Evaluation and Research (CDER).1
In the wake of high-profile safety problems with pharmaceuticals, such as Phen-Fen, Vioxx, and Baycol, FDA needed to address drug safety as observers questioned whether clinical studies were adequately addressing the topic.2 To provide access to medications that can save lives or improve the quality of life for patients and to ensure that these medications are safe calls for a balancing act. Officials at FDA recognize this, stating, "Although FDA has one of the most rigorous preapproval processes in the world, well-conducted, randomized, controlled clinical trials cannot uncover every safety problem, nor are they expected to do so. In most cases, clinical trials aren't large enough, diverse enough, or long enough in duration to provide all the information on a product's performance and safety. In addition, clinical trials are unlikely to reliably detect rare, serious adverse events that occur with long latency or in subpopulations who have not participated in studies."3
When a drug enters the real-world setting, previously unknown safety risks may become evident. Beyond complex medical needs, many specialty drugs treat conditions with small patient populations, adding to the difficulty of obtaining a suitable number of patients for studies. Postmarket safety takes on a new dimension when one considers the permutations of potential drug-drug interactions and the genetic makeup of individual patients. The very mechanisms that are effective in treating a disease can also affect the body's immune system, disrupt one's metabolism, interact with other treatments, and cause unintended effects on other systems. However, the picture can be even more complex for many patients using specialty drugs. Two-thirds of these patients use 5 to 10 different medications in addition to specialty drugs. Another 18% use >10 additional medications.4
REMS programs offer a way to provide safety information to patients and physicians, closely monitor a patient's clinical reaction to the specialty drug, and provide a risk mitigation strategy for drugs that possess both an advanced therapeutic option and a manageable health and safety risk.
SPECIALTY PHARMACIES GAIN PROMINENCE IN CARE
Specialty pharmacies play an important role in the implementation of and compliance with REMS programs. Unlike traditional pharmacies, specialty pharmacies are uniquely designed to dispense and monitor complex high-intensity drug therapies. Because of the complexity of the medical conditions and the medications being used, many drug manufacturers will distribute their products only to specialty pharmacies, which employ specially trained pharmacists and nurses. Specialty drugs often must be shielded from light, need to be transported in temperature-controlled containers, may require unique administration devices, and often are used to treat rare or complex medical conditions, creating a "high-touch" patient care model. Because treatments can cost $6,000 to >$250,000 annually, there is an imperative to reduce waste and ensure proper use of these medications.5
Integrated clinical information networks at specialty pharmacies help with the coordination of care and give clinicians a full picture of the treatments patients receive. The nurses and pharmacists play an important part in educating patients, counseling them about side effects and coordinating care among different medical specialties. Their expertise allows them to serve patients in a way that is not usually available in traditional pharmacies. This level of patient engagement is the very essence of REMS incorporation into the practice of pharmacy care, as feedback about the progress of treatment can be exchanged between patients and the specialty pharmacy's clinical staff.
Specialty pharmacies have grown in tandem with the biotechnology industry, and that trend is likely to continue. The Medco 2009 Drug Trend Report's examination of approximately 130 drugs in the development pipeline demonstrated that approximately 49 of these agents are specialty drugs and approximately 30 will be used for rare or orphan diseases.6 Increasingly, specialty drugs are being used for diseases with larger patient populations, providing another avenue for growth. Also, healthcare payers are seeking to manage medication costs by asking that patients be treated at lower-cost sites of services, which has helped encourage specialty pharmacies to expand into infusion centers.
FDA AND DRUG SAFETY
Although the economic dynamic is shaping how specialty pharmacies conduct business, the clinical dynamic of safety has not changed, nor has it reduced the regulatory demands to ensure patient safety. FDA has been active in identifying and addressing medication safety risks as part of its mission but has had to heighten its efforts after several high-profile recalls. The agency introduced REMS under the FDA Amendments Act of 2007 to replace its Risk Mitigation Action Plan (RiskMAP) program, which gave manufacturers a great deal of responsibility for ensuring medication safety. Some critics of the RiskMAP program charged that it would harm innovation and hinder patient access to medications.7 The REMS program allows drugs to reach the marketplace while requiring a mechanism to provide safety information and ongoing patient monitoring. Under the law establishing REMS, FDA can mandate postmarket studies or continuing clinical trials to address issues of drug safety. In the past, these studies would have been undertaken voluntarily as postmarket commitments.8 REMS also includes an enforcement provision that allows FDA to levy monetary penalties of up to $250,000 per violation (up to $1 million in total).
Since the introduction of REMS in March 2008, approximately one-third of newly approved drugs have been mandated to become part of the program. Some of the drugs affected include certolizumab for the treatment of Crohn's disease and rheumatoid arthritis, tetrabenazine for the treatment of chorea associated with Huntington's disease, and romiplostim for the treatment of chronic immune thrombocytopenic purpura. Before the implementation of the REMS program, drugs already on the market have been affected by FDA, with regulatory actions related to drug safety taken an average of 3.7 years after a medication's launch.9 From January 1995 to June 2007, FDA approved 136 biologics; the agency then required a total of 46 "Dear Healthcare Professional" letters and 19 boxed warnings for these agents.9 REMS has been applied to approved drugs that have a known or potential safety risk, including 6 specialty drugs that were initially part of the RiskMAP program.10 These drugs include ambrisentan, bosentan tablets, eculizumab, lenalidomide, natalizumab, and thalidomide.
Compliance with a REMS program depends on the specific safety issues associated with a drug, providing program flexibility. Risk mitigation strategies may include increased patient monitoring, communication programs, patient registries, restricted distribution, and/or limits on who may prescribe the drugs. In addition to their clinical staff and experience in assisting high-risk patients, specialty pharmacies have developed advanced information systems that are needed to support the documentation and reporting requirements of REMS programs.
Specialty pharmacies are positioned to work with manufacturers in developing best practices for customized drug-delivery services and patient monitoring to meet the unique REMS assigned to a drug. Ongoing safety monitoring and prompt reporting of medical events are key to earlier detection and mitigation of health and safety risks and are components of the specialty pharmacy service model. Specialty pharmacies routinely contact patients and their physicians and can both educate patients about how to mitigate known risks and collect and monitor required health information. In partnership with FDA, the manufacturer and specialty pharmacy can provide a mechanism to balance a biologic's benefits and risks through REMS.
SPECIALTY PHARMACY POSITIONED TO SUPPORT REMS

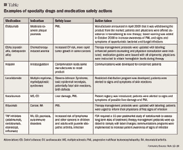
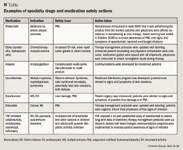
REMS can be viewed as an extension of the specialty pharmacy clinical model; there are many common elements, including advanced patient education and communication, patient monitoring for safe use of the drug, management of drugs with restricted access, early detection and reporting of side effects and adverse medical events, and collection of information for evaluation of REMS effectiveness.
REMS is a flexible program that may include several elements.
Communications. The most basic element associated with REMS is enhancement of patient and physician communication plans. REMS communication plans include information about the safe and appropriate use of a drug and may supplement traditional package inserts. In addition to clinical protocols and patient counseling, REMS communication plans make patients and physicians aware of signs or symptoms that may indicate an adverse medical event. Early identification of these signs or symptoms can help avert an adverse drug event and may also reduce a health plan's exposure to unnecessary healthcare costs. Specialty pharmacy clinical teams work directly with patients and their physicians to provide these early warnings and can help with earlier identification of an adverse medical event.
Clinical monitoring. A REMS requirement may include ongoing patient monitoring of significant laboratory values or other indicators of safe product use. Because of the importance of compliance with a REMS program, the manufacturer may establish a limited distribution network. Monitoring, in tandem with medication guides, will support patient and physician awareness of early symptoms of significant safety risks such as progressive multifocal leukoencephalopathy (PML), Stevens-Johnson syndrome, elevated liver enzymes, and an array of other signposts for adverse drug events.
Some drug categories or individual treatments that may require patient communications and clinical monitoring include erythropoietin-stimulating agents (ESAs), efalizumab, tumor necrosis factor (TNF) inhibitors, and natalizumab. In August 2008, FDA revised the product labeling for ESAs after studies demonstrated that cancerous tumors could grow when the drug dosage is too high during treatment of chemotherapy-induced anemia, effectively negating the benefits of chemotherapy.12,13 Clinicians need to monitor hemoglobin levels to balance the need for transfusions and the safe use of these drugs, including those using the drug for kidney disease. The ESA dose should be individualized to maintain hemoglobin levels within the range of 10 to 12 g/dL. If hemoglobin levels exceed the recommended range, the dose of the ESA needs to be adjusted.13 Studies have demonstrated that patients with hemoglobin levels above the recommended range have a higher risk of death from a cardiovascular event.
Efalizumab is the most recent example of a drug for which a serious safety risk was identified well after the agent was approved and introduced to the market. The immunosuppressant was approved in 2003 for use in chronic moderate-to-severe plaque psoriasis. In October 2008, FDA issued a safety warning for efalizumab that addressed risk of serious infection, including progressive multifocal leucoencephalopathy (PML), a potentially life-threatening infection of the brain associated with this agent. A new medication guide informing patients of the safety risks associated with the drug was issued in March 2009 after 3 of 4 patients afflicted with PML died. In early April 2009, Genentech announced that the company was withdrawing efalizumab from the market and that the drug would no longer be available after June 8, 2009, giving patients time to identify alternative therapy, as efalizumab was not to be abruptly discontinued. Efalizumab was available only from selected specialty pharmacies, which have worked directly with patients and their physicians to transfer patients to new therapies.14
TNF inhibitors, which are used for the treatment of rheumatoid arthritis, Crohn's disease, and other autoimmune conditions, have been available for a decade. The labels for these agents clearly instruct physicians that these drugs should not be used in patients with infection. As part of the monitoring process, patients are also told to inform their physicians about any bruising, bleeding, and paleness or signs of infection that occur during treatment.15,16 Last year, FDA required manufacturers to strengthen the product labels for these agents with more information about the risk of opportunistic fungal infections. Recent studies have demonstrated a connection between TNF inhibitors and the development of lymphoma and other cancers in children and young adults treated for juvenile idiopathic arthritis. As part of a REMS program, the manufacturers of certolizumab pegol are required to undertake a 10-year study to assess the long-term risk of lymphoma and other cancers associated with this agent.
Natalizumab was recalled in 2005, a year after its approval, because of an increased incidence of PML in patients treated with the drug.17 In 2006, the drug returned to the market for the treatment of relapsing forms of multiple sclerosis under a restricted distribution arrangement called the Tysabri Outreach Unified Commitment to Health (TOUCH) Prescribing Program, and this agent was also approved for the treatment of Crohn's disease in 2008.17 The patient-monitoring process has demonstrated an increased risk of liver injury with the drug, and 3 cases of PML were reported last year. The drug has been used by approximately 39,000 patients worldwide, and clinical trials have demonstrated that the agent is effective in preventing relapses of MS.18,19 Patient monitoring has demonstrated that, used as a monotherapy, the drug has a lower safety risk than when it is used concomitantly with other immunomodulatory medications.
Limited access. As part of the REMS program, FDA may require manufacturer-maintained patient registries, dispensing of drugs by certified pharmacies or other certified healthcare settings, and/or prescribing of drugs by specially trained physicians only. Before the introduction of REMS, FDA made recommendations to manufacturers about what elements should be part of a RiskMAP. These recommendations included limiting drug dispensing to pharmacies or practitioners who elected to be specially certified, as well as ensuring that certain reminder systems are in place (eg, consent forms, provider training, data collection, specialized packaging for products).20 These recommendations have been extended to REMS. FDA considers restricted distribution an "element to assure safe use" and an appropriate part of the REMS program.
Certified prescribing/patient registries. Patient registries may be required for drugs with the highest risk profiles, as patients undergoing these treatments must be monitored for early signs and symptoms of safety issues. Clinicians also need to collect clinical information and communicate important safety information to patients and physicians. For several drugs, FDA-mandated RiskMAP safety surveillance programs with mandatory patient registries were in place before the introduction of REMS. Under REMS standards, specialty pharmacies must be certified as meeting a defined level of care in order to dispense these medications and counsel patients. Natalizumab is not the only agent that merits the need for a patient registry and certification; patient registries have also been established for romiplostim, lenalidomide, thalidomide, and bosentan, among others. Patient registries offer a unique opportunity for specialty pharmacy clinical teams to engage patients and provide counseling about side effects and identify earlier signs and symptoms of possible safety risks. From the drug manufacturer's point of view, these registry programs are often marketed with "starter kits" and trademarked names, such as System for Thalidomide Education and Prescribing Safety (STEPS) for thalidomide and Network of Experts Understanding and Supporting Nplate and patients (NEXUS) for romiplostim. Today, special pharmacy plays an integral role in REMS patient engagement and patient registries. Working with the manufacturer, specialty pharmacies ensure that only registered patients receive the REMS drug prescribed by certified practitioners in compliance with the REMS safety program criteria.
In addition to participating in required REMS programs, pharmacists and nurses at specialty pharmacies provide high-touch patient care, reflecting their high degree of expertise in working with complex drug therapies and high-intensity conditions. Specialty pharmacy clinical teams can go beyond the bounds of the REMS program and instruct patients about the proper way to use their therapies, including administration training for drugs that require mixing preparation and self-injection and coordination of services for infused products or for drugs that require special administration devices. By contacting patients or their caregivers with each fill, specialty pharmacies are in a unique position to provide patient education and to reinforce the importance of compliance and continuation of therapy. This advanced level of patient encounters supports REMS-mandated communication and clinical monitoring. Specialty pharmacies also use advanced information systems that support the data collection and reporting requirements of many REMS programs.
Patient registries allow physicians, manufacturers, and pharmacies to manage how a drug is administered. To manage safety risk, certain drugs require ongoing clinical evaluations, such as pregnancy tests, liver function tests, or complete blood counts. Other drugs may require genetic tests to identify patients who are most likely to obtain therapeutic benefit and to reduce the potential of wasting costly medications that deliver no benefit or may even cause patient harm. Sometimes registry programs and specialty pharmacies can also assist patients in navigating their insurance benefits and obtaining reimbursement for many high-cost biologics.
CONCLUSION
As the specialty pharmacy pipeline continues to produce new therapies for many conditions and currently unmet medical needs, it is likely that there will be a larger percentage of drugs that carry a mandated REMS program as part of its FDA approval. REMS programs allow drugs with significant therapeutic benefit and a potentially serious safety issue to be available to patients through careful risk management. The specialty pharmacy model provides the patient contacts, specially trained clinical teams, information systems for documentation and reporting, and infrastructure to support manufacturers and physicians in administering FDA-mandated REMS programs.
A year after the introduction of REMS, it is difficult to say whether the program has succeeded in improving pharmacy safety. FDA's move to require a REMS for ESAs followed an FDA warning in November 2006, just after a New England Journal of Medicine article raised safety concerns about ESAs.21 Despite that quick response, critics are quick to point out that before the implementation of REMS, the drugs had remained on the market for years, despite research that described their risks.
To FDA's credit, REMS goes far beyond simply placing a boxed warning on a drug label. The law gives FDA greater enforcement capability with regard to drug safety, and the fines add an incentive for manufacturers to follow REMS. Some experts in the legal community say a benefit from REMS could be reduced liability from drug safety issues.22 Ronni Fuchs and Michael Carroll wrote in Corporate Counsel that REMS "may one day be your [manufacturers'] best litigation defense," as the process is aimed at increasing warnings to the healthcare and patient communities. REMS represents a departure from RiskMAP, which suggested that the "manufacturer describe the risks to be minimized." The incentives for REMS to be effective are therefore in place.
REMS requires programs to have stated goals, and manufacturers must have ongoing reviews with FDA to determine if the programs are meeting these goals. During a 6-month period ending September 9, 2008, FDA issued 21 letters requiring postmarket studies or clinical trials to address safety issues for various drugs and biologics. FDA changed drug labels 4 times during that period, including the label changes for ESAs and TNF inhibitors mentioned earlier. Eleven of the first 13 approved REMS required medication guides and a timetable for assessment.8
Drug safety is one of the most pressing issues in healthcare today. Beyond the costs of treating adverse drug events, the risk of negatively affecting a patient's well-being adds a degree of complexity to medical practice. Patients needing biologics/specialty drugs are already dealing with medical conditions that are difficult to manage. Their safety needs to be at the forefront of care to prevent unnecessary exposure to risks and complications. REMS programs help to accomplish this with a heightened degree of ongoing clinical monitoring and patient communication. Specialty pharmacies are logical partners in coordination of patient care and REMS compliance. Without a well-managed REMS program, many of today's life-saving drugs might not have been brought to market. Patients benefit from specialty pharmacy services and REMS programs that heighten awareness of safety risks, provide information about therapies, and detect potentially life-threatening adverse events.
Mr Russek is Chief Clinical Officer, Accredo Health Group, Inc., Memphis, Tennessee.
Disclosure Information: The author reports no financial disclosures as related to products discussed in this article.
REFERENCES
1. FDA identifies first steps in requirements for safety plans for certain drugs and biologics [press release]. Washington, DC: FDA; March 27, 2008.
2. Fontanarosa PB, Rennie D, DeAngelis CD. Postmarketing surveillance-lack of vigilance, lack of trust. JAMA. 2004;292:2647–2650.
3. FDA. The Sentinel Initiative: National strategy for monitoring medical product safety. May 2008. http://www.fda.gov/Safety/FDAsSentinelInitiative/ucm089474.htm. Accessed September 14, 2009.
4. Medco Health Solutions, Inc. 2005 survey of patients' PBM claims using specialty drugs. Based upon 241 responses.
5. Medco Health Solutions, Inc. 2008 Drug Trend Report. http://medco.mediaroom.com/file.php/162/2008+drug+trend+report.pdf. Accessed September 14, 2009.
6. Medco Health Solutions, Inc. 2009 Drug Trend Report. http://medco.mediaroom.com/file.php/177/2009+drug+trend+report.pdf. Accessed September 14, 2009.
7. Goldberg R. Restricted treatment: How Congress slows down innovative medicines. Washington Times. March 29, 2007.
8. FDAAA implementation-highlights one year after enactment. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FoodandDrugAdministrationAmendmentsActof2007/ucm083161.htm. Accessed September 14, 2009.
9. Giezen TJ, Mantel-Teeuwisse AK, Straus SM, Schellekens H, Leufkens HG, Egberts AC. Safety-regulated regulatory actions for biologicals approved in the United States and the European Union. JAMA. 2008;300:1887–1896.
10. Identification of drug and biological products deemed to have risk evaluation and mitigation strategies for purposes of the Food and Drug Administration Amendments Act of 2007. Fed Regist. 2008;73:16313–16314.
11. Dickinson, James G. 1 in 8 approval actions call for REMS. Medical Marketing and Media. November 2008;43:17.
12. Procrit [package insert]. Thousand Oaks, CA/Raritan, NJ: Amgen/Ortho Biotech; 2009.
13. Aranesp [package insert]. Thousand Oaks, CA: Amgen; 2009.
14. FDA statement on the voluntary withdrawal of Raptiva from the U.S. market [press release]. Washington, DC: FDA; April 8, 2009.
15. Enbrel [package insert]. Thousand Oaks, CA: Amgen/Wyeth; 2009.
16. Humira [package insert]. North Chicago, IL: Abbott; 2009.
17. FDA. Information for healthcare professionals-natalizumab injection for intraveneous use (marketed as Tysabri)-(8/2008). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126592.htm. Accessed September 14, 2009.
18. Medline Plus. Natalizumab injection. http://www.nlm.nih.gov/medlineplus/druginfo/meds/a605006.html. Accessed September 14, 2009.
19. Stuve O, Cravens PD, Frohman EM, et al. Immunologic, clinical, and radiologic status 14 months after cessation of natalizumab therapy. Neurology. 2009;72:396–401.
20. FDA. Guidance for industry: Development and use of risk minimization action plans. March 2005. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126830.pdf. Accessed September 14, 2009.
21. FDA. FDA public health advisory: Epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). November 16, 2006. http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm124140.htm. Accessed September 14, 2009.
22. Fuchs R, Carroll MR. How to deal with litigation concerns in REMS for biologics. Corporate Counsel. July 9, 2008. http://www.law.com/jsp/cc/PubArticleCC.jsp?id=1202421984742. Accessed September 14, 2009.

Employers Face Barriers With Adopting Biosimilars
March 1st 2022Despite the promise of savings billions of dollars in the United States, adoption of biosimilars has been slow. A roundtable discussion among employers highlighted some of the barriers, including formulary design and drug pricing and rebates.






