
- Safety & Recalls
- Regulatory Updates
- Drug Coverage
- COPD
- Cardiovascular
- Obstetrics-Gynecology & Women's Health
- Ophthalmology
- Clinical Pharmacology
- Pediatrics
- Urology
- Pharmacy
- Idiopathic Pulmonary Fibrosis
- Diabetes and Endocrinology
- Allergy, Immunology, and ENT
- Musculoskeletal/Rheumatology
- Respiratory
- Psychiatry and Behavioral Health
- Dermatology
- Oncology
Therapeutic considerations in managing multiple myeloma
This review highlights treatment guidelines, disease staging criteria, and new therapeutic approaches to MM, including an overview of supportive care and emerging pipeline agents.



Key Points
Abstract
Management strategies for multiple myeloma (MM) have continued to evolve as the result of innovative treatment modalities and efficacy data used in establishing new standards of care, including new combination therapy. Most commonly diagnosed in the growing elderly population, MM continues to be one of the most costly cancers because of the need for stem cell transplant and medication used in initial and relapse stages, as well as in supportive care. The National Comprehensive Cancer Network and the International Myeloma Workgroup maintain guidelines that outline diagnosis strategies, disease staging, and treatment algorithms according to disease severity of MM and transplant eligibility of the patient. This review highlights these new treatment approaches to MM, including an overview of supportive care and emerging pipeline agents. With the recent advancements in MM, the need for more cost-effective strategies in managing this condition is heightened. (Formulary. 2008;44:204–213.)

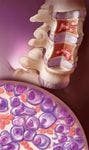
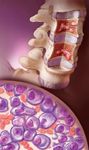



Myeloma tumor burden may be approximated by the ratio of existing myeloma cells and the amount of monoclonal proteins (M-proteins) produced by these myeloma cells. Found in the blood and/or urine, M-proteins are fragments of or whole immunoglobulins that often serve as clinical markers of MM. The most commonly produced M-protein is immunoglobulin G (IgG) (52%), followed by IgA (21%), and rarely, IgD and IgM.5,6 Abnormal M-proteins consisting of only the light chain portion of the immunoglobulin structure, also known as Bence Jones proteins, occur in 16% of cases. Serum free light chain (FLC) assay is used when M-protein level has not reached disease threshold levels (ie, serum M-protein ≥1 g/100 mL or urine ≥200 mg/24 h); data obtained from the serum FLC assay can be used for prognosis and predicting response to therapy.4

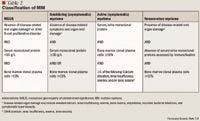
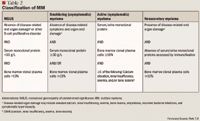
Although MM remains an incurable disease, ongoing growth in the development of viable therapeutic options provides a promising outlook for the management of this disease. The initiation of pharmacotherapy for MM is dependent on the degree of disease progression as evidenced by laboratory and clinical data. Staging systems used to categorize disease severity of active MM have been published both nationally and internationally. The International Staging System (ISS) and the Durie-Salmon staging system for MM were established in 2005 and 1975, respectively.10,11 These staging systems both use 3-stage severity classifications that provide laboratory criteria for each stage but differ in the complexity, reproducibility, and actual criteria constituting each stage. According to Durie-Salmon, Stage I is defined as hemoglobin >10 g/dL, normal serum calcium or serum calcium ≤12 mg/dL, low monoclonal component (IgG <5 g/dL, IgA <3 g/dL, and/or Bence Jones proteins <4 g/24 h), and normal bone structure or solitary bone plasmacytoma only. Based on ISS, Stage I is defined as serum albumin ≥3.5 g/dL and serum beta2-microglobulin <3.5 mg/L; beta2-microglobulin is a surface protein in many blood cells, including white blood cells. For both Durie-Salmon and ISS, Stage II is defined as not meeting criteria established for Stage I or III. For Durie-Salmon, Stage III is defined as ≥1 of the following: hemoglobin <8.5 g/dL, serum calcium >12 mg/dL, high monoclonal component (IgG >7 g/dL, IgA >5 g/dL, and/or Bence Jones proteins >12 g/24 h), and/or advanced lytic bone lesions (scale 3). For ISS, Stage III is defined as serum level of beta2-microglobulin ≥5.5 mg/L. Durie-Salmon includes a subclassification to reflect renal function, in which "A" reflects serum creatinine <2 mg/dL and "B" correlates to serum creatinine ≥2 mg/dL).
Having been validated against the Durie-Salmon staging system and in patients worldwide, the ISS is currently the most widely used staging system. Shortcomings of the ISS include its inability to differentiate a higher stage due to tumor burden versus renal failure, as well as its applicability only in patients who have already been diagnosed with MM and who are taking conventional rather than new or investigational therapies for MM.4 As such, investigators recommend the concurrent use of both the ISS and Durie-Salmon staging systems. Recent studies have suggested that other factors may serve as more reliable, independent prognostic factors than established MM staging systems in patients with active MM disease.12,13
There has been progressive development in risk stratification for MM through pharmacogenomic research, which is aimed at estimating survival and treatment response. Ongoing research links MM to specific chromosomal abnormalities detected by immunophenotyping, chromosomal analysis by conventional karyotyping, and fluorescence in situ hybridization (FISH).14 Information provided by MM genetics and gene expression profiling will provide insight into factors that should be considered for accurate detection of cases associated with poor prognosis and for prediction of therapeutic response and design of personalized therapy.4,14 Some investigators recommend that all patients diagnosed with MM be screened for DNA hypodiploidy, immunoglobulin heavy chain (IgH) translocations (ie, t[4; 14], t[14; 16]), chromosome 13 deletion, and p53 deletion to aid in appropriate disease management for patients who would be deemed likely to be nonresponders to standard therapy with high-dose melphalan and stem cell transplantation.14
PATHOPHYSIOLOGY
In normal human physiology, B lymphocytes or B cells are produced by hematopoietic stem cells in the bone marrow and migrate to various parts of the body through the bloodstream. Triggered by the body's humoral immune response to antigens, certain B cells differentiate into plasma cells, which produce and secrete large quantities of immunoglobulin (ie, antibodies); these immunoglobulins attach to the antigens that had originally elicited their proliferation. The normal plasma cells remain in the bone marrow.



Survival, growth, and differentiation of the remaining normal plasma cells rely heavily on certain extracellular matrix proteins and cell components in the bone marrow.15,16 MM cells interact with bone marrow stromal cells (BMSCs) through their intracellular and vascular cell adhesion molecules, which are upregulated by kappaB nuclear factor; this interaction results in a cascade of events in the bone marrow microenvironment, including downstream secretion of cytokines (ie, interleukin-6, insulin-like growth factor 1, and vascular endothelial growth factor) and antiapoptotic proteins, both of which further promote a cycle of adhesion and cytokine secretion.16,17 Continued research aimed at creating a better understanding of the bone marrow microenvironment will contribute to advancing our knowledge of MM pathogenesis and new targets for treatment.
PHARMACOLOGIC TREATMENT OF MULTIPLE MYELOMA
Determination of treatment for MM is dependent on diagnostic criteria, risk stratification, and initial symptomatology. For instance, asymptomatic patents with MM do not require treatment, as treatment will not result in clinical benefit. However, these patients should be monitored for disease progression, with evaluation every 3 to 6 months.8 Conversely, symptomatic disease requires treatment consisting of a hematopoietic stem cell transplant (HSCT) and chemotherapy.






Since the 1960s, patients ineligible for HSCT have been treated with glucocorticoids or alkylating-based therapy such as melphalan and prednisone (MP). This regimen has been demonstrated to improve median survival to approximately 3 years.20 Recently, studies investigating the addition of thalidomide to this regimen have demonstrated an improved median overall survival (OS) of approximately 5 years.17,21 This has earned the melphalan, prednisone, and thalidomide (MPT) regimen classification as the preferred initial therapy in patients with MM who are not candidates for HSCT. The basis of this designation was the result of data collected in several clinical trials. Palumbo et al22 initially compared MPT to MP in 255 patients and reported PR plus CR rates of 76% with MPT versus 47.6% with MP; CR plus near CR (nCR) rates were 28% and 7.2%, respectively. In addition, the 2-year event-free survival rates were 54% with MPT and 27% for MP (P=.0006). Event-free survival rates were not significantly different at 3 years (80% vs 64%; P=.19).
In a second study, 447 patients were randomized to 1 of 3 arms: MPT, MP, or intermediate-dose melphalan (100 mg/m2) followed by HSCT.23 Results demonstrated a higher rate of at least PR with MPT and intermediate-dose melphalan compared with MP (76% vs 65% vs 35%, respectively; P<.0001 for both comparisons vs MP). Compared with MP, CR rates were also higher with MPT (P=.0008) and with intermediate-dose melphalan (P<.0001) (2% vs 13% vs 18%, respectively). Median progression-free survival (PFS) was significantly better with MPT versus MP (27.5 mo vs 17.8 mo; P<.0001) and versus intermediate-dose melphalan (27.5 mo vs 19.4 mo; P=.0002). OS was also greater with MPT versus MP (51.6 mo vs 33.2 mo; P=.0006) and versus intermediate-dose melphalan (51.6 mo vs 38.3 mo; P=.027).
Palumbo et al24 randomized a total of 331 patients to treatment with MP or MPT. Results demonstrated improved CR, very good partial response (VGPR), and PR rates with MPT versus MP (15.6% vs 3.7%; P<.001; 29.3% vs 11%; P<.001; and 68.9% vs 47.6%; P<.001, respectively).
An alternative to MP and MPT is the combination of bortezomib, melphalan, and prednisone (MPB). Bortezomib is a novel proteasome inhibitor that is approved by FDA as an initial treatment for MM. A phase 1/2 clinical trial of MPB in 60 patients newly diagnosed with MM demonstrated an overall response rate (CR plus PR) of 89%. At a median follow-up of 16 months, 91% of patients were free of disease progression, and 2-year OS rate was 86%.25 In addition, the 3-year OS rate was 85%.26 This combination regimen was also studied versus MP in 682 patients.27 Time to progression among patients receiving MPB was 24 months versus 16.6 months among patients receiving MP (P<.001). MPB was associated with a PR rate of 71% versus 35% with MP (P<.001); CR rates were 30% and 4%, respectively (P<.001), and the median duration of the response was 19.9 months in patients treated with MPB and 13.1 months in patients treated with MP. Adverse events were consistent with previously reported profiles of toxic effects associated with bortezomib and MP. Combined with previous data, these results led to the FDA approval of MPB for the initial treatment of MM.27
The National Comprehensive Cancer Network (NCCN) qualifies its recommendations using an alphanumerical system that describes both the level of evidence and the degree of consensus of the organization in support of the recommendations.8 Most optimal, category 1 correlates to high-level evidence and uniform NCCN consensus. Categories 2A and 2B both are correlated with low-level evidence but differ in that category 2A is associated with uniform consensus, whereas category 2B is associated with nonuniform consensus (ie, no major disagreement). Category 3 reflects major disagreement among NCCN panel members in their support of the recommendation.
According to NCCN, regimens deemed as category 1 for primary induction for patients who are not eligible for a transplant include MPB and MPT, as described above. Treatment options designated as category 2A choices by NCCN include MP, vincristine plus doxorubicin plus dexamethasone (VAD), thalidomide plus dexamethasone, and dexamethasone alone. The remaining regimens, including lenalidomide plus low-dose dexamethasone and liposomal doxorubicin plus vincristine plus dexamethasone (DVD), are category 2B options.8 These therapies should be reserved for patients who cannot tolerate or have contraindications to category 1 treatment options.
Although newer therapies have been demonstrated to be more efficacious, they are not devoid of side effects. For instance, thalidomide is associated with a higher rate of thromboembolism and peripheral neuropathy than melphalan-based regimens. As a result, deep vein thrombosis prophylaxis must be considered along with thalidomide treatment. Prophylaxis is also warranted with lenalidomide plus dexamethasone therapy. Bortezomib may increase the incidence of infection, especially herpes zoster reactivation. As a result, prophylactic antiviral medication is recommended.
Treatment of transplant candidates. Compared with chemotherapy alone, induction therapy along with subsequent HSCT increases CR rates and prolongs OS by up to 1 year in previously untreated patients with MM.8 As a result, HSCT plus induction therapy is considered a category 1 recommendation for the treatment of MM. In patients who are candidates for autologous HSCT, induction chemotherapy is the initial component of treatment. Induction chemotherapy is administered for 2 to 4 months before stem cell collection in an effort to reduce the number of tumor cells within the bone marrow and peripheral blood, decrease symptoms, and minimize end-organ damage. Among transplant candidates, the preferred induction regimen is unknown. However, exposure to myelotoxic therapies including alkylating agents such as melphalan are generally avoided to minimize the risk of compromising stem cell reserves before harvest.28 As a result, the initial chemotherapy regimen chosen for transplant candidates differs from that chosen for patients who are not transplant candidates.
Historically, dexamethasone alone (category 2A) was considered the standard regimen for induction therapy. However, recent studies evaluating its use in combination with thalidomide, bortezomib, and lenalidomide have reported higher response rates with combination therapy. Currently, thalidomide with dexamethasone (category 2A) is considered a standard induction regimen. Efficacy of this regimen was demonstrated in a phase 3 Eastern Cooperative Oncology Group (ECOG) trial that compared thalidomide plus dexamethasone with dexamethasone alone in 207 previously treated patients.29 Overall response rate was greater with combination therapy than with monotherapy (63% vs 41%, respectively; P=.0017). Another randomized, double-blind, placebo-controlled trial confirmed these results. In this study, 470 patients received thalidomide plus dexamethasone or placebo plus dexamethasone. Patients treated with thalidomide plus dexamethasone had significantly higher rates of overall response (63% vs 46%; P<.001) and longer time to progression of disease (22.6 mo vs 6.5 mo; P<.001).30
Another combination therapy that has demonstrated promising results is lenalidomide plus dexamethasone (category 2B). An analogue of thalidomide, lenalidomide is approved by FDA for the treatment of relapsed or refractory MM but has also been studied as an induction therapy. A phase 2 study examined combination therapy with clarithromycin, lenalidomide, and dexamethasone in treatment-naïve patients; this treatment resulted in improved CR, nCR, PR, and objective response (at least PR) rates (38.89%, 13.89%, 16.67%, and 90.3%, respectively).31 In addition, a phase 2 trial of lenalidomide plus dexamethasone reported that 91% of patients newly diagnosed with MM achieved at least PR, 6% achieved CR, and 32% met criteria for VGPR and nCR.32
Bortezomib has also demonstrated significant clinical benefit as part of an induction therapy. In an open-label, phase 2 study, patients newly diagnosed with MM were given bortezomib in combination with dexamethasone (category 2B).33 This regimen resulted in a CR of 21%, a VGPR of 10%, and an overall response rate of 66%. Bortezomib is associated with a high rate of peripheral neuropathy. In an effort to decrease toxicity yet preserve clinical efficacy, a study was conducted using an alternating schedule of bortezomib as induction therapy followed by autologous HSCT.34 Bortezomib was administered every other cycle rather than every cycle. With this regimen, 65% of patients demonstrated at least PR (PR, 42.5%; CR, 12.5%; VGPR, 10%). Reported toxicity was low, with 22.5% of patients reporting grade 1 peripheral neuropathy and 2.5% reporting grade 2 peripheral neuropathy. Investigators have also conducted a retrospective comparison of bortezomib plus dexamethasone versus VAD as induction therapy.35 Objective response rate (at least PR) before stem cell transplant was not significantly different between the groups. However, high-quality response rate (at least VGPR) before stem cell transplant was significantly higher with bortezomib plus dexamethasone treatment versus VAD (66.7% vs 34.2%, respectively; P=.006). After stem cell transplant, there was no difference between the treatment groups in PFS or OS.
Other therapies under investigation that are categorized as 2B include bortezomib plus dexamethasone plus thalidomide, bortezomib plus doxorubicin plus dexamethasone, and bortezomib plus lenalidomide plus dexamethasone. When using DVD (category 2A), a central line must be placed to prevent vincristine-associated neurotoxicity. Alternatively, thalidomide plus dexamethasone and lenalidomide plus dexamethasone are administered orally.
MANAGEMENT OF RELAPSED DISEASE
Despite the development of promising treatment options, MM is not a curable disease. Unfortunately, most patients will develop resistance to therapy with a subsequent relapse. When this occurs, salvage therapy is warranted. Determination of an appropriate salvage regimen is dependent on a number of factors, including initial therapy regimen used and duration of response to that therapy. Treatment options for relapsed disease include HSCT, a rechallenge of a previous chemotherapy regimen, or a trial of a new chemotherapy regimen. Many of the same chemotherapy regimens that are used as initial therapy may be used as salvage therapy. Category 1 salvage therapies include bortezomib, bortezomib plus liposomal doxorubicin, and lenalidomide plus dexamethasone. A number of other category 2A and 2B salvage therapies exist, but this discussion is beyond the scope of this manuscript.
SUPPORTIVE CARE
Management of active MM often requires adjunctive pharmacotherapy. The NCCN identifies 7 complications of MM requiring pharmacotherapeutic care, including bone disease, hypercalcemia, hyperviscosity, anemia, infection, renal dysfunction, and coagulation issues.8 Most of these complications result from the normal cells being crowded out by myeloma cells. Supportive care for these conditions is discussed in the following sections.
Bone disease. Occurring in 80% to 85% of patients with MM, bone pain and pathologic fractures from lytic bone lesions are commonly reported.6,8 These bone lesions exist because of the large quantities of M-protein that inhibit osteoblasts and stimulate osteoclasts involved in bone formation and bone resorption (ie, destruction), respectively. Patients with MM exhibiting bone-related complications, even osteopenia, should be treated with a bisphosphonate.8 Consensus guidelines support continued bisphosphonate therapy in the presence of active bone disease rather than indefinite treatment.36,37 Controversy exists regarding the appropriate duration of bisphosphonate therapy based on limited long-term data; expert opinions vary relative to factors warranting discontinuation of therapy as well as use of a reduced dosing scheduled (ie, every 3 mo).38 Based on evolving published literature, recommendations for the appropriate initiation and duration of bisphosphonate therapy in MM may change over time and require re-evaluation.39
Examples of bisphosphonates that are approved by FDA for treating bone loss in patients with MM include intravenous pamidronate and zoledronic acid. Because of a lower risk for drug-related osteonecrosis of the jaw, pamidronate is the preferred bisphosphonate over zoledronic acid, according to NCCN.8 However, there is no available evidence to suggest that switching patients from zoledronic acid to pamidronate is warranted at this time.37 Available outside the United States, oral clodronate represents another bisphosphonate demonstrated to be effective in MM, according to published literature.36,37
Low-dose radiation (10–30 Gy) may be used conservatively as palliative care for uncontrolled bone pain, including pain associated with fracture and cord compression.8 An annual skeletal survey is recommended for monitoring bone disease, but densitometry is not essential. Other surgical interventions, including kyphoplasty, may be considered for pain alleviation and strengthening of damaged bones.
Hypercalcemia. Directly related to M-protein overproduction, increased osteoclast activity is correlated with an increase in serum calcium levels. Hypercalcemia is observed in 13% of patients with MM.6 Treatment options for managing hypercalcemia include use of furosemide and hydration measures, steroids, bisphosphonates, and/or calcitonin.8
Hyperviscosity/coagulation issues. Patients with MM are at risk for deep vein thrombosis and venous thromboembolism because of both the hyperviscosity and hypercoagulability state of the patient's blood caused by the overproduction of M-proteins. Although patients with protein S deficiency and elevated coagulation factors trend towards an increase in prothrombotic risk in MM, the clinical significance of this risk cannot be predicted on the basis of a specific laboratory value at this time.40 Symptomatic blood hyperviscosity may require plasma exchange.8 Chemotherapy should be resumed only when the viscosity of the blood is normalized. Management strategies are aimed at preventing complications such as mucosal hemorrhage, vision impairment, and neurologic issues. Patients taking thalidomide or lenalidomide with dexamethasone for the treatment of MM are at a greater risk for coagulation issues and should therefore also receive anticoagulation prophylaxis with aspirin, warfarin, or low molecular weight heparin.8
Anemia. When myeloma cells multiply, other cell components within the bloodstream are compromised. Anemia and bleeding can result from decreased red blood cells and platelets, respectively. Primary anemia occurs in 70% to 72% of patients with MM.6,41 Anemia can also worsen with kidney dysfunction and can occur secondary to chemotherapy. Using erythropoiesis-stimulating agents (ESA) in these patients can reduce transfusion requirements. In light of recent data supporting an increased mortality risk associated with ESAs, treatment with darbepoetin alfa and epoetin alfa should be considered only in anemic patients receiving radiation or chemotherapy.42,43 The duration of this treatment should be limited to the treatment period regardless of target hemoglobin levels (ie, <12 g/dL).
Immunodeficiency. Susceptibility to infections is heightened in patients with MM, as their production of white blood cells is reduced because of the inability of the myeloma cells to produce normal antibodies. To minimize the potential for infections, patients with MM should be vaccinated against Streptococcus pneumoniae, Haemophilus influenzae, and influenza.8,44 In addition, a patient who is taking any MM drug that may cause a treatment-related infection should receive appropriate prophylactic therapy. Examples of such therapy include herpes zoster prophylaxis with bortezomib or Pneumocystis carinii pneumonia (PCP), herpes, and antifungal prophylaxis with high-dose dexamethasone.8 However, patients with a history of recurrent, life-threatening infections should receive treatment with intravenous immunoglobulin (IVIG).
Nephropathy. Reported in 19% of patients with MM, renal insufficiency occurs when the kidney is not able to regulate blood composition because of excessive M-protein and calcium levels.6 Excessive quantities of Bence Jones proteins may lead to proteinuria and further nephropathy; in these patients, high urine output (3 L/d) is recommended.9 Adjunctive treatment should be aimed at managing dehydration, hyperuricemia, hypercalcemia, and any other electrolyte imbalance. By either lowering the dose (ie, for drugs requiring a constant blood level) or prolonging the dosing interval (ie, for drug efficacy that is dependent on peak levels), healthcare professionals should consider renal dosing for all drugs that are renally excreted. Of note, patients with MM also receiving bisphosphonate therapy should be monitored for worsening renal function. Nonsteroidal anti-inflammatory drugs (NSAIDs), intravenously administered contrast media, and other nephrotoxic agents should be avoided in patients with renal impairment.
EMERGING THERAPIES
In light of the many investigational agents currently being studied for the treatment of MM, the concept of terminal prognosis once associated with MM may soon be replaced by a prognosis that is more in line with that of a chronic disease.44 The International Myeloma Foundation maintains the "Myeloma Matrix," which serves to continuously track the progress (from preclinical to phase 3 clinical trials) of pipeline agents and combination therapies being studied for the management of MM.45 The following section explores selected novel agents in the pipeline according to their respective innovative targets in MM pathophysiology, particularly as relating to the MM cells, the bone marrow microenvironment, cell surface receptors, and/or a combination of these. According to preliminary data, these selected agents have demonstrated promising antitumor effects in patients with refractory or relapsed MM.
Salinosporamide A (NPI-0052) and carfilzomib (PR-171). Similar to bortezomib, salinosporamide A is a proteasome inhibitor but with distinct properties that make this drug more than another "me-too" agent. Salinosporamide A differs from bortezomib in its chemical structure, mechanism of action, safety profile (ie, less toxicity), and kinetic profile (ie, ability to elicit a quicker cellular response in vitro). Orally administered salinosporamide A is being studied in a dose-escalation phase 1 clinical trial in patients with refractory and relapsed MM.46 Injectable carfilzomib, another novel proteasome inhibitor, is being studied in multiple phase 1b and 2 trials as monotherapy or in combination with bortezomib or lenalidomide and dexamethasone in patients with relapsed or refractory MM and/or with evidence or varying degrees of renal function.47,48 Synergistic apoptotic effect through combination therapy with bortezomib and other proteasome inhibitors may be possible in the future because of each agent's unique properties.49
Tanespimycin (KOS-953) and retaspimycin (IPI-504). Not unique to MM, heat shock protein 90 (Hsp90) inhibitors are being investigated for the treatment of hematologic and solid tumors. The role of Hsp90 is primarily related to maintaining conformational stability for cellular functionality of key proteins, especially cancer-related proteins or cell-signaling proteins that are overexpressed in cancer and crucial in cancer cell proliferation and survival.44 The Hsp90 inhibitors bind to and inhibit the cytosolic chaperone functions of Hsp90. The Hsp90 inhibitors tanespimycin and retaspimycin are being studied in patients with relapsed and/or refractory MM.50 A phase 2/3 study is being conducted to evaluate tanespimycin compared with bortezomib in a dose-finding study in patients with relapsed and refractory MM who have previously undergone therapy with bortezomib and lenalidomide. A phase 3 study evaluating PFS associated with tanespimycin plus bortezomib compared with bortezomib alone in patients with MM in first relapse has been developed but is currently suspended as of March 2009.51 A safety study has been completed for retaspimycin in patients with MM; phase 1, 2, and 3 trials are being conducted to evaluate this agent for use in other cancers. Hsp90 inhibitors may offer another treatment option as monotherapy or as adjunctive anticancer agents for patients who do not respond to proteasome inhibitors.
Other pipeline agents not discussed in this manuscript include farnesyl transferase inhibitors, arsenic trioxide, Akt/protein kinase B inhibitors (eg, perifosine), histone deacetylation (HDAC) inhibitors, rapamycin inhibitors, vascular endothelial growth factor inhibitors, insulin-like growth factor 1 receptor inhibitors, tumor necrosis factor (TNF)-related apoptosis-inducing ligand, and a number of monoclonal antibodies (eg, tocilizumab), among others.16,17,44 To improve efficacy while minimizing toxicity, combination therapeutic regimens of 2 or 3 agents are being studied for the treatment of MM.52 Though they are beyond the scope of this manuscript, it is important to also note that novel agents are being developed to better control the secondary complications of MM, such as bone disease.53
ECONOMIC CONSIDERATIONS
The introduction of newer therapies for the treatment of MM has resulted in a significant increase in cost of overall therapy. One of the primary reasons for this phenomenon is that the newer therapies are recommended as add-on therapies as opposed to replacement therapies. In addition, the majority of therapies recommended by the NCCN are combinations of ≥2 medications because of reported efficacy outcomes. For example, 1 cycle of chemotherapy can range from $33,000 to $71,000 for bortezomib-, thalidomide-, or lenalidomide-based regimens.54 As the gold-standard therapy for remission in MM, autologous HSCT is also costly, with an estimated total expenditure of $20,000 to $60,000.54 In addition, the cost of induction therapy, which routinely consists of 3 to 4 cycles of high-dose chemotherapy, must be considered. Support therapy for secondary complications of MM also contributes to this rising cost of overall MM care. The rates (ie, 28.8%) and incremental cost (ie, $63,455) of metastatic bone disease were demonstrated to be highest for patients with MM compared with patients with other cancer types.55 Alternative methods for managing anemia in light of economic and safety concerns have been reviewed.56,57 The financial burden of caregivers for patients with MM must also be considered. All these factors bring forth viable cost-related concerns surrounding the management of MM. With the aging population (ie, aged ≥65 y) projected to be 80 million by 2040, it is even more important to proactively evaluate the economic effect of new therapeutic approaches and devise cost-effective strategies for managing MM today.55
CONCLUSION
From the first confirmed case of MM in 1844 and the first successful treatment with melphalan in 1962, pharmacotherapeutic options available for the management of MM today have become far more sophisticated, with even more promising agents in the development pipeline. Panel members of the NCCN published updated clinical practice guidelines for the management of MM in February 2009 to reflect current treatment recommendations. Initial treatment for MM is dependent on risk stratification and initial clinical picture. If a patient is not a candidate for the gold-standard therapy with HSCT, proteasome inhibitors and immunomodulators have demonstrated positive outcomes when used in a combination regimen of 2 to 3 agents. Supportive care is typically required for patients experiencing secondary complications, such as bone disease, caused by progression of the MM. Growing evidence supporting improved clinical response with newer agents may change current recommendations, which recognize transplant as the standard of care. New targets for therapy are being investigated, including agents aimed at the bone marrow microenvironment. Current research on pharmacogenomics in MM will offer better outcomes through use of personalized medicine. Continued evaluation of the clinical and economic effect of these new treatment modalities used for the comprehensive management of MM is critical because of the aging population and greater costs that we will need to face in the future.
Dr Lee is associate director for professional development, University of Massachusetts Medical School, Commonwealth Medicine–Clinical Pharmacy Services, Shrewsbury. Dr De Bellis is associate director of clinical services, University of Massachusetts Medical School, Commonwealth Medicine–Clinical Pharmacy Services.
Disclosure Information: The authors report no financial disclosures as related to products discussed in this article.
REFERENCES
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96.
2. Ries LAG, Melbert D, Krapcho M, et al (eds). SEER cancer statistics review, 1975-2005, National Cancer Institute. http://seer.cancer.gov/csr/1975_2005/. Accessed June 16, 2009.
3. Brenner H, Gondos A, Pulte D. Expected long-term survival of patients diagnosed with multiple myeloma in 2006–2010. Haematologica. 2009;94:270–275.
4. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9.
5. Nair B, Waheed S, Szymonifka J, et al. Immunoglobulin isotypes in multiple myeloma: Laboratory correlates and prognostic implications in total therapy protocols. Br J Haematol. 2009;145:134–137.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003:78:21–33.
7. The International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757.
8. The NCCN clinical practice guidelines in oncology: Multiple myeloma (version 2.2009). National Comprehensive Cancer Network. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed June 16, 2009.
9. Barosi G, Boccadoro M, Cavo M, et al; Italian Society of Hematology; Italian Society of Experimental Hematology; Italian Group for Bone Marrow Transplantation. Management of multiple myeloma and related disorders: Guidelines from the Italian Society of Hematology (SIE), Italian Society of Experimental Hematology (SIES), and Italian Group for Bone Marrow Transplantation (GITMO). Haematologica. 2004;89:717–741.
10. Greipp P, San Miquel J, Durie B, et al. International staging system for multiple myeloma [erratum in J Clin Oncol. 2005:23:6281]. J Clin Oncol. 2005;23:3412–3420.
11. Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842–854.
12. Perosa F, Minoia C, Favoino E, Prete M, Dammacco F. Staging multiple myeloma patients with active disease using serum levels of beta-2m-free HLA class I heavy chain together with IgM or platelet count. Blood Cells Mol Dis. 2009;42:71–76.
13. Bataille R, Grenier J, Sany J. Beta-2-microglobulin in myeloma: Optimal use for staging, prognosis, treatment-a prospective study of 160 patients. Blood. 1984;63:468–476.
14. Stewart AK, Fonseca R. Prognostic and therapeutic significance of myeloma genetics and gene expression profiling. J Clin Oncol. 2005;23:6339–6344.
15. Jagannath S. Pathological underpinnings of multiple myeloma progression. J Manag Care Pharm. 2008;14:S7–11.
16. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–598.
17. Katzel JA, Hari P, Vesole DH. Multiple myeloma: Charging toward a bright future. CA Cancer J Clin. 2007;57:310–318.
18. Rajkumar SV. Determination of appropriate initial therapy in patients with multiple myeloma. UpToDate database. http://www.utdol.com/utd/index.do. Accessed June 16, 2009.
19. Durie BGM, Harousseau JL, Miguel JS, et al; International Myeloma Working Group. International uniform response criteria for multiple myeloma [errata in Leukemia. 2006;20:2220 and 2007; 21:1134]. Leukemia. 2006;20:1467–1473.
20. Alexanian R, Haut A, Khan AU, et al. Treatment for multiple myeloma. Combination chemotherapy with different melphalan dose regimens. JAMA. 1969;208:1680–1685.
21. Palumbo A, Bertola A, Musto P, et al. Oral melphalan, prednisone, and thalidomide for newly diagnosed patients with myeloma. Cancer. 2005; 104:1428–1433.
22. Palumbo A, Bringhen S, Caravita T, et al; Italian Multiple Myeloma Network; GIMEMA. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: Randomised controlled trial. Lancet. 2006; 367:825–831.
23. Facon T, Mary JY, Hulin C, et al; Intergroupe Francophone du Myélome. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): A randomised trial. Lancet. 2007; 370:1209–1218.
24. Palumbo A, Bringhen S, Liberati AM, et al. Oral melphalan, prednisone, and thalidomide in elderly patients with multiple myeloma: Updated results of a randomized controlled trial. Blood. 2008;112:3107–3114.
25. Mateos MV, Hernandez JM, Hernandez MT, et al. Bortezomib plus melphalan and prednisone in elderly untreated patients with multiple myeloma: Results of a multicenter phase 1/2 study. Blood. 2006;108:2165–2172.
26. Mateos MV, Hernandez JM, Hernandez MT, et al. Bortezomib plus melphalan and prednisone in elderly untreated patients with multiple myeloma: Updated time-to-events results and prognostic factors for time to progression. Haematologica. 2008;93:560–565.
27. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917.
28. Boccadoro M, Palumbo A, Bringhen S, et al. Oral melphalan at diagnosis hampers adequate collection of peripheral blood progenitor cells in multiple myeloma. Haematologica. 2002; 87:846–850.
29. Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR; Eastern Cooperative Oncology Group. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: A clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431–436.
30. Rajkumar SV, Rosinol L, Hussein M, et al. Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J Clin Oncol. 2008;26:2171–2177.
31. Niesvizky R, Jayabalan DS, Christos PF, et al. BiRD (Biaxin [clarithromycin]/Revlimid [lenalidomide]/dexamethasone) combination therapy results in high complete- and overall-response rates in treatment-naive symptomatic multiple myeloma. Blood. 2008;111:1101–1109.
32. Rajkumar SV, Hayman SR, Lacy MQ, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood. 2005;106:4050–4053.
33. Harousseau J, Attal M, Leleu X, et al. Bortezomib plus dexamethasone as induction treatment prior to autologous stem cell transplantation in patients with newly diagnosed multiple myeloma: Results of an IFM phase II study. Haematologica. 2006;91:1498–1505.
34. Rosinol L, Oriol A, Mateos MV, et al. Phase II PETHEMA trial of alternating bortezomib and dexamethasone as induction regimen before autologous stem-cell transplantation in younger patients with multiple myeloma: Efficacy and clinical implications of tumor response kinetics. J Clin Oncol. 2007;25:4452–4458.
35. Eom HS, Min CK, Cho BS, et al. Retrospective comparison of bortezomib-containing regimens with vincristine-doxorubicin-dexamethasone (VAD) as induction treatment prior to autologous stem cell transplantation for multiple myeloma. Jpn J Clin Oncol. 2009;39:449–455.
36. Berenson JR, Hillner BE, Kyle RA, et al; American Society of Clinical Oncology Bisphosphonates Expert Panel. American Society of Clinical Oncology clinical practice guidelines: The role of bisphosphonates in multiple myeloma. J Clin Oncol. 2002;20:3719–3736.
37. Lacy MQ, Dispenzieri A, Gertz MA, et al. Mayo clinic consensus statement for the use of bisphosphonates in multiple myeloma. Mayo Clin Proc. 2006;81:1047–1053.
38. Durie BGM. Use of bisphosphonates in multiple myeloma: IMWG response to Mayo Clinic consensus statement [letter]. Mayo Clin Proc. 2007;82:516–517.
39. Drake MT, Clarke BL, Khosla L. Bisphosphonates: Mechanisms of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032–1045.
40. Auwerda JJ, Sonneveld P, de Maat M, Leebeek FW. Prothrombotic coagulation abnormalities in patients with newly diagnosed multiple myeloma. Haematologica. 2007;92:279–280.
41. Durie BG, Kyle RA, Belch A, Bensinger W, Blade J, Boccadoro M, et al; Scientific Advisors of the International Myeloma Foundation. Myeloma management guidelines: A consensus report from the Scientific Advisors of the International Myeloma Foundation [erratum in Hematol J. 2004;5:285]. Hematol J. 2003;4:379–398.
42. Bennett CL, Silver SM, Djulbegovic B, et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. JAMA. 2008;299:914–924.
43. The NCCN clinical practice guidelines in oncology: Cancer- and chemotherapy-induced anemia (version 3.2009). National Comprehensive Cancer Network. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed June 16, 2009.
44. Schwartz RN. Vozniak M. Current and emerging treatments for multiple myeloma. J Manag Care Pharm. 2008;14:S12–S18.
45. Myeloma matrix (10/20/08). International Myeloma Foundation. http://myeloma.org/main.jsp?source=link&source_link_id=981&type=article&tab_id=3&menu_id=31&id=735. Accessed June 16, 2009.
46. Chauhan D, Singh A, Velankar M, et al. Distinct dynamic profiles for NPI-0052- and bortezomib-induced apoptosis in multiple myeloma [abstract]. Blood. 2006;108:970a.
47. Phase 2 study of carfilzomib in relapsed and refractory multiple myeloma. Clinicaltrials.gov/. http://clinicaltrials.gov/ct2/show/NCT00511238?term=carfilzomib&rank=4. Accessed June 29, 2009.
48. Phase 1b multicenter study of carfilzomib with lenalidomide and dexamethasone in relapsed multiple myeloma. Clinicaltrials.gov/. http://clinicaltrials.gov/ct2/show/NCT00603447?term=carfilzomib&rank=3. Accessed June 29, 2009.
49. Chauhan D, Singh A, Brahmandam M, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111:1654–1664.
50. Siegel D, Mazumder A, Borello I, et al. Update on phase I clinical trial of IPI-504, a novel, water-soluble Hsp90 inhibitor, in patients with relapsed/refractory multiple myeloma [abstract]. Blood. 2006;108:1022a.
51. A study of tanespimycin (KOS-953) in patients with multiple myeloma in first relapse (BMS TIME-1). Clinicaltrials.gov/. http://clinicaltrials.gov/ct2/show/NCT00546780?term=tanespimycin+myeloma&ranl=3. Accessed June 16, 2009.
52. Lonial S, Cavenagh J. Emerging combination treatment strategies containing novel agents in newly diagnosed multiple myeloma. Br J Haematol. March 13, 2009 [Epub ahead of print].
53. Esteve FR, Roodman GD. Pathophysiology of myeloma bone disease. Best Pract Res Clin Haematol. 2007;20:613–624.
54. Cook R. Economic and clinical impact of multiple myeloma to managed care. J Manag Care Pharm. 2008;14:S19–S25.
55. Schulman KL, Kohles J. Economic burden of metastatic bone disease in the U.S. Cancer. 2007;109:2334–2342.
56. Pizzi LT. Economic considerations in a changing anemia environment. Am J Kidney Dis. 2008;52:S29–S33.
57. Birgegard G. Managing anemia in lymphoma and multiple myeloma. Ther Clin Risk Manag. 2008;4:527–539.

Employers Face Barriers With Adopting Biosimilars
March 1st 2022Despite the promise of savings billions of dollars in the United States, adoption of biosimilars has been slow. A roundtable discussion among employers highlighted some of the barriers, including formulary design and drug pricing and rebates.






